Definition
Neuronal ceroid lipofuscinosis (NCL) is the general name for a family of at least eight genetically separate neurodegenerative disorders that result from excessive accumulation of lipopigments (lipofuscin) in the body's tissues. These lipopigments are made up of fats and proteins. Their name comes from the word stem lipo-, which is a variation on "lipid" or "fat", and from the term pigment, used because the substances take on a greenish-yellow color when viewed under an ultraviolet light microscope. These lipofuscin materials build up in neuronal cells and many organs, including the liver, spleen, myocardium, and kidneys.
Kufs disease is an adult type of inherited neurodegenerative neuronal ceroid lipofuscinosis, where lipopigments accumulate specifically in the nervous tissue, causing progressive motor and cognitive deficits. The particularity of Kufs disease is that it very rarely affects vision, or does not affect it at all.

Fig. 1 Types of neuronal ceroid lipofuscinosis
Kufs Disease (Type A and B); Oct 2012
Neuronal Ceroid Lipofuscinosis; Sep 2013
Neuronal Ceroid Lipofuscinosis; Jan 2014
Epidemiology
Collectively, all forms of NCL affect an estimated 1 in 100.000 individuals worldwide. NCLs are more common in Finland, where approximately 1 in 12.500 individuals have the condition. Kufs disease is thought to represent 1.3-10% of all NCLs.
How common is Kufs Disease? (From Kufs Disease); Sep 2013
Signs and Symptoms
Kufs disease is a very rare disorder marked initially by progressive weakness with diminished muscle coordination, seizures, rapid involuntary jerky movements (chorea) and, rarely, blindness. This disorder can be inherited as either a dominant or recessive trait and is usually slowly progressive.
Neurological symptoms of Kufs disease can resemble mental illness. Confusion, stupor or psychotic behavior may mark the onset, leading to mental retardation and generalized convulsions. These symptoms are due to excess pigment in fat (lipofuscins) that accumulate in the brain.

Fig. 2 Infantile neuronal ceroid lipofuscinosis
People with Kufs disease may present with a skin disorder involving dryness, roughness and/or scaliness (ichthyosis vulgaris). This condition arises as a result of excess production and/or retention of keratin (one of the principal components of skin).
The clinical manifestations of Kufs disease usually appear approximately at age 30, but the range of onset age spans from adolescence to late adulthood. The signs and symptoms of Kufs disease worsen over time, and affected individuals usually survive about 15 years after the disorder begins.
Kufs Disease; May 2008
Molecular Basis
The molecular basis of Kufs disease is not completely understood, whereas a series of genes accounting for most of the childhood-onset forms of neuronal ceroid lipofuscinosis (NCL) have been identified.
Mutations in the CLN6 or PPT1 gene are related with Kufs disease type A and mutations in the DNAJC5 or CTSF gene are related with Kufs disease type B. Most of the proteins or enzymes produced from these genes are involved in breaking down proteins or clearing unneeded materials from cells.
Mutations in the CLN6, PPT1, DNAJC5, or CTSF gene usually reduce the activity of the gene or impair the function of the protein or enzyme produced from the gene. In many cases, these mutations cause incomplete breakdown of certain proteins and other materials. These materials accumulate in the lysosome, forming fatty substances called lipopigments. In Kufs disease, these accumulations occur in nerve cells in the brain, resulting in cell dysfunction and eventually cell death. The progressive death of neurons leads to the signs and symptoms of Kufs disease.
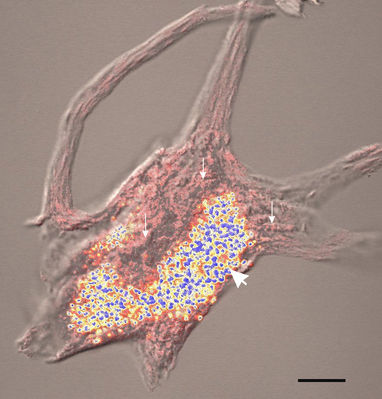
Fig. 3 Retention of Nissl bodies and lipfuscin by extracted motor neurons isolated from human lumbar spinal cord and stained with methylene blue. The pink structures (small arrows) are Nissl bodies and the blue and yellow structures are lipofuscin granules (large arrow)
Some people with either type of Kufs disease do not have an identified mutation in any of these four genes. In these individuals, the cause of the condition is unknown.
Mechanisms of Lysosomal Degradation
Lysosomes are the cell's waste disposal system and can digest some compounds. They contain acid hydrolase enzymes that break down waste materials and cellular debris. Acid hydrolases may be nucleases, proteases, glycosidases, lipases, phosphatases, sulfatases and phospholipases and make up the approximately 50 degradative enzymes of the lysosome that break apart biological matter. These enzymes require an acid pH, so the interior of the lysosomes has a pH 4.8.
Acid hydrolases come into contact with the substances to degrade by means of a process called phagocytosis or autophagy depending on the origin of these substances.
- In phagocytosis, large particles (such as a bacterium or a virus) are incorporated (endocitated) from the cell. Also in this case a vesicle forms and it is derived from the fusion of the cell membrane (endosome) with the lysosome.
- Autophagy is the biological process of degradation of cellular proteins by lysosomal vesicles derived from the membrane of the endoplasmic reticulum. The closure of these membranes determines the formation of the autophagosome. In the next steps the membrane of the autophagosome fuses with the late endosome or lysosome, forming the autolysosome. In autophagy CLN6 (ceroid-lipofuscinosis neuronal 6) protein is involved: it regulates the transport of certain proteins and fats from the endoplasmic reticulum to lysosomes.

Fig. 4 Lysosomes in phagocytosis and autophagy
Between the acid hydrolases there are two proteins whose modifications are involved in the development of some neuronal ceroid lipofuscinosis, such as Kufs Disease: cathepsin F (CTSF protein) and palmitoyl-protein thioesterase 1 (PPT-1).
- Cathepsins are a group of proteases (enzymes that degrades proteins). There are approximately a dozen members of this family, which are distinguished by their structure, catalytic mechanism, and which proteins they cleave. One of them, cathepsin F, is involved in Kufs disease: this protein is a cysteine proteinase, so that it cleaves after a cysteine residue.
- Palmitoyl-protein thioesterase 1 (PPT-1) is a small glycoprotein involved in the catabolism of lipid-modified proteins during lysosomal degradation. This enzyme removes thioester-linked fatty acyl groups such as palmitate from cysteine residues.
Lysosome; Feb 2014
Cathepsin; Jan 2014
Phagocytosis and Autophagy; 2000
Types of Kufs Disease
Two types of Kufs disease have been described: type A and type B. The two types are differentiated by their genetic cause, pattern of inheritance, and certain signs and symptoms.
- Kufs disease type A is characterized by a combination of seizures and uncontrollable muscle jerks (myoclonic epilepsy), a decline in intellectual function (dementia), impaired muscle coordination (ataxia), involuntary movements such as tremors or tics and speech difficulties (dysarthria).
- Kufs disease type B shares many features with type A (dementia, ataxia and extrapyramidal signs), but it is distinguished by changes in personality and is not associated with myoclonic epilepsy or dysarthria.
What is Kufs Disease? (From Kufs Disease); Sep 2013
The CLN6 gene (ceroid-lipofuscinosis, neuronal 6, late infantile, variant) is located on the long arm of chromosome 15 at position 23 (15q23). It provides instructions for making a protein that likely regulates the transport of certain proteins and fats from the endoplasmic reticulum to lysosomes. Based on this function, the CLN6 protein appears to help in the process of ridding cells of materials they no longer need.
It is unclear how mutations in the CLN6 gene are involved in the buildup of lipopigments and the features of Kufs disease. Research suggests that CLN6 gene mutations that cause Kufs disease allow enough functional protein to be produced so that signs and symptoms of the disorder do not develop until adulthood. The presence of other proteins that compensate for the lack of CLN6 protein could also play a role in the appearance of symptoms in adulthood.
CLN6 gene; Sep 2013
Kufs disease, the major adult form of neuronal ceroid lipofuscinosis, caused by mutations in CLN6; May 2011
The PPT1 gene (palmitoyl-protein thioesterase 1) is located on the short arm of chromosome 1 at position 32 (1p32). It provides instructions for making an enzyme called palmitoyl-protein thioesterase 1. This enzyme has been found in lysosomes and removes long-chain fatty acids from proteins; a decrease in the normal amount of palmitoyl-protein thioesterase 1 slows the breakdown of fatty acids and proteins. The materials accumulate over time into lipopigments in the lysosomes of nerve cells in the brain.

Fig. 5 Palmitoyl-protein thioesterase 1 (PPT1)
At least four mutations in the PPT1 gene have been found to cause Kufs disease type A. These mutations change single amino acids or create a premature stop signal in the instructions used to make palmitoyl-protein thioesterase 1. Also the PPT1 gene mutations allow enough functional protein to be produced so that signs and symptoms of Kufs disease do not develop until later in life.
PPT1 gene; Aug 2013
Palmitoyl-Protein Thioesterase 1 (PPT1); Sep 2013
Kufs disease type B, caused by mutations in the CTSF or DNAJC5 gene, has an autosomal dominant pattern of inheritance, which means one copy of the altered gene in each cell is sufficient to cause the disorder. Most cases of Kufs disease type B occur in people with no history of the disorder in their family.
The CTSF gene (cathepsine F) is located on the long arm of chromosome 11 at position 13 (11q13). It provides instruction for making an enzyme called cathepsin F. Cathepsin F, which has been found in many types of cells and in lysosomes, acts as a protease. By cutting proteins apart, cathepsin F can break proteins down, turn on proteins, and regulate self-destruction of the cell (apoptosis).

Fig. 6 Cathepsin F (CTSF)
At least five mutations in the CTSF gene have been found to cause Kufs disease type B. Most of these mutations change single amino acids, resulting in a cathepsin F protein with reduced function. This decrease of function likely slows the normal breakdown of proteins and other materials.
CTSF gene; Sep 2013
DNAJC5 gene (Heat Shock Protein 40 homolog, subfamily C, member 5) belongs to a family of genes called DNAJ and is located on the long arm of chromosome 20 at position 13.33 (20q13.33). It provides instructions for making a protein called cysteine string protein alpha (CSPα). This protein has been found in the brain, where it plays a role in the transmission of nerve impulses by ensuring that nerve cells receive signals.
At least two mutations in the DNAJC5 gene have been found to cause Kufs disease type B: the first mutation replaces the amino acid leucine with arginine at position 115 in the CSPα protein; the second deletes the amino acid leucine at position 116 in the protein.
These DNAJC5 mutations occur in one copy of the gene in each cell and lead to the production of an altered protein that does not associate with the synaptic vesicle, resulting in impaired nerve impulse transmission.
DNAJC5 gene; Sep 2013
Diagnosis
The diagnosis of Kufs disease is based on clinical history, physical and neurologic exam.
Clinical findings include myoclonus, ataxia, Parkinsonism, chorea, spasticity and cognitive impairment. Lipopigment inclusions, most frequently described in neuron cytoplasm, have been found in extra neural tissues, including vascular smooth muscle cells, Schwann cells and sweat gland epithelial cells, so biopsy of the skin or other tissues, examined under the electron microscope, represent a method of diagnosis. In Kufs disease, the deposits have a characteristic fingerprint-type pattern.
Electroencephalogram may be useful to document seizures.
Genetic testing may show mutations in the CLN or PPT1 genes, but it is not required for diagnosis.
Kufs’ disease: diagnostic difficulties in the examination of extracerebral biopsies; Mar 2009
Treatment
There is no treatment to cure or slow down the progression of Kufs disease. Anticonvulsive drugs are helpful to control seizures and myoclonic jerking. Physical, speech and occupational therapies can help individuals function for as long as possible. Experimental therapies, including gene therapy, are used for NCL disorders.
Treatment, from Neuronal Ceroid Lipofuscinosis (Wikipedia); Jan 2014
Salvatico Erica
Rosso Roberta