An Historical view. . . . .

DEFINITION
Pseudohermaphroditism is a term used to describe the condition of individuals who have phenotypes and secondary sex characteristics that are different from what would be expected on the basis of their karyotype and gonads. It differentiates from true hermaphroditism since in this case there is the presence of both male and female gonads in a single individual.
Pseudohermaphrodites may present a complete form of the opposite phenotype, but there are also intermediate conditions in which external sex organs have an ambiguous aspect.
There are two different types of pseudohermaphroditism:
• male pseudohermaphroditism is a condition in which individuals with a XY karyotype and testes appear with a complete or partial female phenotype.
• female pseudohermaphroditism is the presence of complete or partial male phenotypes in individuals with a XX karyotype and ovaries.
Both these conditions result from disorders of sex development during fetal life, due to many different problems involving the presence or the absence of androgens, estrogens and their corresponding receptors in the target tissues. Without these hormones and their receptors, the internal and external sex organs, and psyche, will not develop as expected.
Disease definition, Britannica, Robert D. Utiger
EPIDEMIOLOGY
The estimated incidence of male pseudohermaphroditism is between 1/20.000 and 1/99.000 live male births.
Female pseudohermaphroditism has a prevalence of 1 case per 16.000 population.
MALE PSEUDOHERMAPHRODITISM
PATIENT RISK FACTORS and PATHOGENESIS
This condition is due to hypoandrogenism in XY individuals, that is a lack in the presence or in the action of testosterone and dihydrotestosterone.
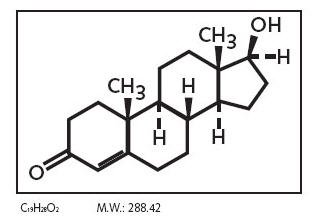
The action of testosterone is normally indispensable in male fetus for the development of primary sexual characteristics that include the creation of internal and external male genitalia.

In particular masculinization of the external genitalia (penis, penile urethra, and scrotum), as well as the prostate, are dependent on the action of the metabolite Dihydrotestosterone, in witch Testosterone is converted by the 5-alpha reductase enzyme. This enzyme is produced in many tissues, in both males and females, especially in the reproductive tract, testes and ovaries but also skin, seminal vesicles, prostate and epididymis. There are three isoenzymes of 5-alpha reductase witch present different distribution with age: isoenzymes 1 and 2 are more represented during fetal life and after birth while isoenzyme 3 is ubiquitously expressed in adults. Dihydrotestosterone is much more powerful than testosterone and, like testosterone, it needs to bind the androgen receptor AR in order to regulate the transcription of target genes involved in development of male genitalia.
In addiction to this, direct effects of Testosterone are needed during the male fetal life to promote the development of Wolffian ducts that will subsequently develop into the epididymides, vasa deferentia, and seminal vesicles. Testosterone is essential both to induce testes descend into the scrotum and to promote spermatogenesis by acting directly on sertoli cells witch under its stimulation produce grow and survival factors for spermatozoons.
Furthermore Testosterone is responsible of the development of secondary sexual characteristics occurring after puberty: it promotes protein synthesis that leads to an increased musculature and an increased quantity of body hair, the larynx becomes larger resulting in a deeper voice and adipose tissue is redistributed and sets in the abdominal area.

Lastly Testosterone has the primary role to lead to a masculinization of the brain during fetal development. Although is known that testosterone also acts in the brain indirectly trough the action of estradiolo in witch it is converted by the enzyme aromatase, actually in humans the direct role of testosterone is essential. During the critical periods of male fetus development, the presence of testosterone is in fact fundamental for masculinization of sexual dimorphic areas of the brain and subsequently consolidation of male gender. The importance of Aromatase in this process is nowadays to confirm even if its presence in the central nervous system leads to presume that this enzyme might be involved in the masculinization of certain areas of the brain. Aromatase is a member of the cytochrome P450 family (p450 19A), encoded by the gene CYP19, located on chromosome 15q21. Its name is due to the fact that it aromatizes the A steroidal ring of androgens converting them into estrogens.
In conclusion it can be affirmed that Testosterone, in its direct or indirect form, is essential for development of both physical and mental characteristic of male gender.
Deficit in the presence and in the action of these hormones is due to different conditions that can be divided into three main categories:
• androgen resistance: it is the main cause of male pseudohermaphroditism and it leads to the Androgen Insensitivity Syndrome
• deficit in the production of testosterone
• deficit in the production of dihydrotestosterone
Main cause of male pseudohermaphroditism:
Androgen insensitivity syndrome
Androgen insensitivity syndrome (AIS) consists in the partial or complete inability of the cell to respond to androgens. This condition of unresponsiveness compromises the masculinization of male genitalia in the developing fetus, as well as the development of male secondary sexual characteristics at puberty. Individuals affected by AIS present phenotypes that range from a normal male aspect with mild spermatogenic defect and reduced secondary terminal hair, to a full female phenotype, despite the presence of a Y-chromosome.

Androgen Insensitivity Syndrome can be divided into three categories based on the degree of genital masculinization:
• Complete androgen insensitivity syndrome (CAIS) is a condition of complete inability of the cell to respond to androgens. The resistance of cell toward testosterone and its metabolite dihydrotestosterone prevents the complete masculinization of male genitalia in the developing fetus and the development of male secondary sexual characteristics at puberty. All affected individuals are phenotypically female, despite the presence of an XY kariotype.

• Partial androgen insensitivity syndrome (PAIS) consists in the partial inability of the cell to respond to androgens. In this case the degree of androgen insensitivity of cells of individuals with a 46 XY karyotype can partially, but not completely, prevent the masculinization of the genitalia. This results in series of ambiguous phenotypes in witch the genitalia are partially masculinized and partially feminized.
• Mild androgen insensitivity syndrome (MAIS) is a condition of mild inability of cells to respond to androgens. It can be defined as a reduction in sensitivity toward androgens that is sufficient to impair spermatogenesis and development of secondary sexual characteristics at puberty in males. However it does not affect genital differentiation and the clinical phenotype associated with MAIS is a normal male phenotype with mild spermatogenic defect and reduced secondary terminal hair.
Androgen insensitivity syndrome, from Wikipedia, the free encyclopedia

The condition of androgen insensitivity is due in the great majority of cases to mutations in the androgen receptor (AR)
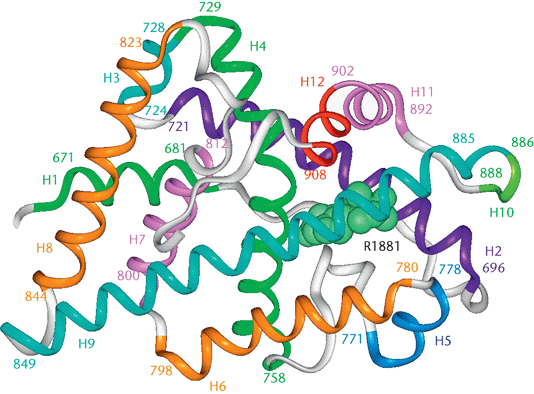
The human androgen receptor is a monomeric protein with a molecular mass of 100–110kD, encoded by a gene located on the proximal long arm of the X chromosome (locus Xq11-Xq12).The protein coding region consists of approximately 2.757 nucleotides (919 codons) divided into eight exons (from 1 to 8 or from A to H). Introns vary in size between 0.7 and 26 kb. The androgen receptor protein consists of several functional domains: the transcription-regulation domain, that is the amino / NH2-terminal domain, the DNA-binding domain and the steroid-binding domain, that is also called the carboxyl-terminal ligand-binding domain. There is also a small hinge region between the DNA-binding domain and ligand-binding domain. The N-terminal domain, which has a modulatory function, is encoded by exon 1 (1586 bp); the DNA-binding domain is encoded by exons 2 and 3 (152 and 117 bp, respectively); the ligand-binding domain is encoded by five exons, (from 131 to 288 bp). This receptor contains 11 α-helices and two short β-turns. Helices H1 and H3 form one face of the ligand-binding domain, while helices H4 and H5, the first β-turn, and helices H8 and H9 form the central part of the structure. Helices H6, H7, H10, and H11 constitute the second face of the ligand-binging domain. H5, the N-terminal region of H3 and the C-terminal region of H10 and H11 form the main part of the hydrophobic ligand-binding pocket where androgens set. When the ligand enters the pocket, H12 is repositioned and stabilizes the androgen position. The very end of the C-terminal region of the ligand-binding domain forms the second β-turn (next to H8 and H10), which further stabilize the H12 conformation. The transcription-regulation domain is an hydrophobic groove formed by the C-terminal region of H3, loop 3-4, H4, and H12. The DNA-binding domain recognizes and binds to the androgen responsive element ARE through two zinc fingers structures.

Unbound AR is mainly located in the cytoplasm and associated with a complex of heat shock proteins (HSPs) that interact with the ligand-binding domain. As a consequence of testosterone bond, AR goes through a series of conformational changes: the heat shock proteins dissociate from AR and the transformed AR homodimerizes. It undergoes phosphorylation and then translocation to the nucleus, which is mediated by the nuclear localization signal. Translocated receptor binds to the androgen response element (ARE), which is characterized by consensus sequences of six nucleotides ( 5′-TGTTCT-3′ ) spaced by three random nucleotides and is located in the promoter or enhancer region of AR gene targets. The activation of AR-regulated gene expression needs the recruitment of other transcription co-regulators (co-activators and co-repressors) and the transcriptional machinery. All of these complicated processes are initiated by the ligand-induced conformational changes in the ligand-binding domain.
AR mutations
Mutations of the Androgen Receptor can be distributed throughout the entire gene, even if the five exons constituting the ligand-binding domain are mainly prone to alterations. More than 400 AR mutations have been reported in the AR mutation database and many others continue to appear. Inheritance is typically maternal since the gene is set in X chromosome and it follows an X-linked recessive pattern. Individuals with a XY karyotype will always express the mutant gene and present androgen insensitivity syndrome since they only have one X chromosome. On the other hand XX carriers will be minimally affected: genetic females in fact have two X chromosomes, and thus have two AR genes. A mutation in one of the two AR genes results in a minimally affected, fertile, female carrier. Some carriers have been noted to have slightly reduced body hair, delayed puberty, and tall stature, presumably due to anomalous X-inactivation. A female carrier has 50% of possibility to pass the mutated AR gene to her children. If the affected child is a genetic female, she too will be a carrier. A genetic female with mutations in both AR genes could theoretically result from the union of a fertile man with androgen insensitivity syndrome and a female carrier, condition that is quite impossible since men affected by AIS are rarely fertile.
In the 30% of the time the AR mutation is not inherited but it appears as an ex novo mutation that can be the result of a germ cell mutation in the gonads of the parents or a mutation in the fertilized egg itself. It has to be noticed that not every mutation of the AR gene results in androgen insensitivity. However in the great majority of cases alterations of this gene are pathological. Mutations in the steroid-binding domain usually affect androgen binding affinity; mutations in the hinge region compromise nuclear translocation; mutations in the DNA-binding domain affect dimerization and binding to target DNA while mutations in the transactivation domain induce the impossibility of target gene transcription regulation. Some mutations can even compromise more than one functional domain: in fact a mutation in one functional domain can have deleterious effects on another by altering the way in which the domains interact. The steroid binding domain, that is the carboxyl- terminal domain, is particularly vulnerable to the effects of a premature stop codon or framing error, since it occurs at the end of the gene.
The Androgen Receptor Gene Mutations Database
Complete Androgen Insensitivity Syndrome is always associated with an AR mutation that completely destroy Androgen Receptor function: target cells do not respond to testosterone or dihydrotestosterone. An AR mutation is found in more than 95% of patients affected by CAIS and 30% of these present ex novo mutations.
Partial Androgen Insensitivity Syndrome is often due to missense mutations in the Androgen Receptor gene witch result in variable degrees of dysfunction Of this receptor. However an AR mutation is found in only approximately 20% of patients with PAIS.
Since some individuals with CAIS or PAIS do not have any AR mutations (about 5% of women with CAIS, as well as 27-72% of individuals with PAIS) other causes have to be researched.
Complete and Partial Androgen Insensitivity Syndrome can also be the result of a deficit in the molecules needed for the transmission of a transactivating signal from the N-terminal domain of the normal androgen receptor. In particular the deficit of a specific coactivator protein interacting with the transactivation domain of the androgen receptor has been proved to be one of the other possible cause of androgen resistance.
In addition to this there is evidence that Partial Androgen Insensitivity Syndrome can be also caused by a mutant steroidogenic factor-1 (SF-1) protein. SF1 protein, encoded by the NR5A1 gene, is a transcription factor and a coactivator of many genes that promote the transcription of steroidogenic enzymes, cholesterol transporters, steroidogenesis-stimulating hormones and steroidal receptors. It is expressed by urogenital ridge cells and is required for the development of the adrenal glands and gonads. It also coactivates SRY gene ( that encode for sex-determining region Y protein) leading to subsequential activation of SOX9 and AMH. Mutations in SF1 gene can lead to a misregulation of genes encoding for steroidal receptors like the androgen receptor.
Pathophysiology of Androgen insensitivity syndrome, from Medscape Reference, Author: Christian A Koch, MD, PhD, FACP, FACE; Chief Editor: Stephen Kemp
Other genetic RISK FACTORS
Deficit in the production of Testosterone
Deficit in testosterone synthesis leads to the complete lack of this hormone and of its metabolite dihydrotestosterone, that results in male hermaphrodites phenotypically identical to patients with CAIS or severe PAIS.
However in this case the molecular causes of female phenotype are different since they involve deficit in enzymes that provide to testosterone synthesis.
Two of the most important involved enzymes are Cytochrome P450c17 and 17β-Hydroxysteroid dehydrogenases (17β-HSD).

Cytochrome P450c17 and 17-hydroxylase deficiency
Cytochrome P450 17α-hydroxylase/17,20-lyase (CYP17) is a microsomal enzyme that can be found in leydig cells, ovarian follicles, and the adrenal zonae fasciculata and reticularis. It is the product of the cytochrome P45017 alpha gene (CYP17A1), set on chromosome 10 and consisting of 8 exons and 7 introns. Cytochrome P450c17 presents two distinct activities: it finctions at the same time as a 17α-hydroxylase and a 17,20-lyase. It is essential for the biosynthesis of sex steroid precursors since it mediates 17α-hydroxylation of pregnenolone or progesterone to obtain 17α-OH pregnenolone or 17α-OH progesterone and also mediates cleavage of the c17,20 bond of these compounds to lead to the synthesis of dehydroepiandrosterone (DHEA) and androstenedione that are fundamental for the production of testosterone. Obviously this enzyme is basic also for estradiol and cortisol synthesis.
P450c17 defects affect both adrenal and gonadal steroid production. Mutations of this gene cause different degrees of 17-hydroxylase deficiency, that can be isolated to 17,20 lyase deficiency or may involve both of its enzymatic activities.
More than 80 different genetic mutations of the CYP17A1 gene have been described and many of these occur more commonly in different populations.
17-β-Hydroxysteroid dehydrogenase III deficiency
17β-Hydroxysteroid dehydrogenases is another group of enzymes involved in steroidogenesis. They are oxidoreductases which catalyze the dehydrogenation of 17-hydroxysteroids: in particular isoenzyme 3, predominantly expressed in the testis, catalyses the interconversion of androstenedione into testosterone.
Mutations in 17β-HSD III gene can lead to complete or partial lack of this enyme that result in a syndrome called 17-beta-hydroxysteroid dehydrogenase deficiency, an autosomal recessive disorder.
Also in this case people affected by this deficiency may present either ambiguous external genitalia or complete female external genitalia, due to different degree of deficit of testosterone and subsequently deficit in masculinization.
Defects in androgen biosynthesis causing 46,XY disorders of sexual development
17-Hydroxylase Deficiency Syndrome
Author: J Paul Frindik, MD, FACE; Chief Editor: Stephen Kemp, from Medscape Reference
5-alpha-reductase type 2 deficiency
5-alpha-reductase type 2 enzyme is mainly located in fetus epididymides, in seminal vesicles, in the prostate and in the external genitalia. It consists of 254 amino acids and it is encoded by a gene located in chromosome 2p. 33 different mutations of this gene have been found in patients affected by deficit of 5-alpha-reductase type 2 enzyme. However correlation between the severity of the syndrome and a particular gene defect has not been observed. 5-alpha-reductase type 2 deficiency is an autosomal recessive sex-limited condition.
Mutations of this enzyme result in the impossibility to convert testosterone into the more physiologically active dihydrotestosterone essential to provide masculinization of male external genitalia in uterus. As a result individuals with mutant 5-alpha-reductase type 2 are born with ambiguous genitalia.
Pathophysiology of 5-Alpha-Reductase Deficiency, from Medscape Reference, Author: Jill E Emerick, MD; Chief Editor: Stephen Kemp
5α-reductase 2 deficiency, Thieme Medical Publishers 333 Seventh Avenue, New York, NY 10001, USA
SYMPTOMS
Classification and Phenotypical Symptoms of male pseudohermaphroditism
The phenotypes associated with the androgen insensitivity syndrome can be classified by the Quigley scale which consists of seven classes depending on the different degree of genital masculinization. Grade 1 includes individuals whose external genitalia are fully masculinized, condition that correspond to mild androgen insensitivity syndrome. Grade 6 and 7 includes individuals whose external genitalia are fully feminized: while grade 6 indicates the presence of secondary terminal hair appearing after puberty, grade 7 indicates the secondary terminal hair absence. Grade 6 and 7 of the Quigley scale are attributed to individuals affected by complete androgen insensitivity syndrome. Grades 2-3-4-5 represent different degrees of genital partial masculinization: these ambiguous conditions are typical of individuals affected by partial androgen insensitivity syndrome.
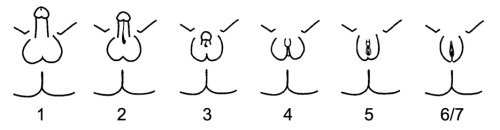
Also patients with 5-alpha-reductase type 2 deficiency can be classified on the basis of different degrees of masculinization of the external genitalia, using the Prader scale. This scale presents five different stages of virilization: stage 1 is the complete female phenotype, while stage 5 correspond to complete masculinization.

Individuals with complete androgen insensitivity, as well as individuals with severe deficit in testosterone production are born with a complete female phenotype, despite having a 46, XY karyotype. They have no signs of genital masculinization.

First symptoms of these conditions do not appear until puberty, which may be slightly delayed, but it is normal except for the absence of menses and diminished or absent secondary terminal hair like axillary hair.
Female external genitalia are normal, although labia and clitoris are sometimes underdeveloped. The vaginal depth is typically shorter than unaffected women.
Although phenotypical appearance the gonads in these women are not ovaries but testes. In fact testes develop during fetal life by an androgen-independent process involving the SRY gene on the Y chromosome. The Sex determining Region Y gene is set in Yp11.3 and encodes a transcription factor that is a member of the SOX gene family, the testis determining factor (TDF), which is responsible for the development of undifferentiated gonads into testes.
These testes may be located intra-abdominally, at the internal inguinal ring and they may even herniate into the labia majora. They can produce Testosterone even if it cannot be used since the presence of a mutant androgen receptor. Instead testosterone produced by testes is aromatized into estradiolo, which effectively feminizes the body and even guarantees well developed breasts.
Immature sperm cells in the testes do not mature, since testosterone is required in order for spermatogenesis to complete.
In patients affected by CAIS or severe deficit in testosterone production Wolffian structures (the epididymides, vasa deferentia, and seminal vesicles) are absent like the prostate and the external male genitalia. On the other hand in male pseudohermaphrodites affected by deficit of 5-alpha-reductase Wolffian structures differentiation is normal, presenting development of seminal vesicles, vasa differentia, epididymides, and ejaculatory ducts since this process of differentiation is dependent on the presence of direct testosterone. In this case the prostate is small and rudimentary.
In all pseudohermaphrodite men, the Müllerian system (the fallopian tubes, uterus, and upper portion of the vagina) typically regresses because of the presence of anti-Müllerian hormone. AMH, encoded by the AMH gene set in 19p13.3, is a dimeric glycoprotein which inhibits the development of the Müllerian ducts. It is secreted by Sertoli cells of the testes during embryogenesis of the fetal male under stimolation of different factors like SF1, SRY and SOX9. Because of AMH presence there is no presence of fallopian tubes, a cervix or a uterus and vagina is a blind ending vagina.
Moreover individuals who have a severe lack of testosterone develop not only as phenotypical women but they have also no gender problems since their brains result feminized by the absence of testosterone effects during fetal development.
The result is that these individuals present a male genotype XY and at the same time they are phenotypically female: they can be considered as women since even their brains are feminized and they have all female gender characteristics. They have female sexual orientation and they feel themselves as women. In fact even if the "true" sex of an individual is determined by the internal organs, the external phenotype and the orientation of the brain determined the "perceived" sex of an individual and its own gender.
The condition of feminization of the brain do not presents in patients affected by 5-alpha-reductase type 2 deficiency, since the presence of direct testosterone guarantees masculinization of the sexual dimorphic areas of central nervous system.
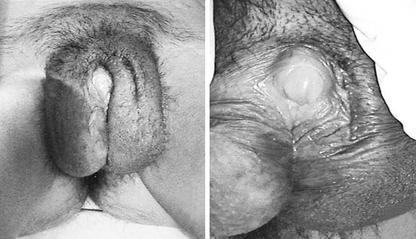
Individuals affected by Partial Androgen Insensitivity Syndrome or who have partial deficit of the synthesis of testosterone are born with an ambiguous phenotype: the phallic structure ranges from a penis of diminished size to a slightly enlarged clitoris while the gonads are always testes.
Wolffian structures can be partially or fully developed: this condition depends on the degree of androgen resistance affecting the individual. The prostate is typically small or impalpable.
Patients who present predominantly male phenotypes may present different degrees of genital undermasculinization that can include micropenis, chordee, bifid scrotum and hypospadias. Condition of impotence and anejaculation may be common.
Otherwise patients with predominantly female phenotypes may present labial fusion and clitoromegaly.
Individuals with mild androgen insensitivity syndrome are born phenotypically male, according to their XY karyotype. They have fully masculinized genitalia and their problems are mostly related to the condition of infertility (oligospermia or azoospermia), decreased secondary terminal hair, high pitch of voice. The external male genitalia (penis, scrotum, and urethra) are normal, as well as internal genitalia, including Wolffian structures (the epididymides, vasa deferentia, and seminal vesicles) and prostate. However testicular volume can be diminished due to the condition of infertility.
COMPLICATIONS
Each form of androgen absence or insensitivity is associated with infertility since spermatogenesis strictly requires the presence of testosterone.
Lack of testosterone is also associated with a decreased bone mineral density. It have been hypothesized that the decreased bone mineral density may be a consequence of gonadectomy and subsequent inadequate estrogen supplementation. This is possible because as long as the testes are not removed they continue to produce testosterone that it peripherally converted into estradiolo that provides to maintain bone mineral density. However it has also been hypothesized that this deficiency is directly attributable to the role of androgens in bone mineralization.
In addition to this, individuals affected by cryptorchidism have an increased risk for gonadal tumors, due for example to germ cell malignancy. The risk of malignant germ cell tumors increases with age from 3.6% at 25 years to 33% at 50 years.
Patients with female phenotypes may present vaginal hypoplasia, that is a form of underdevelopment or incomplete developmet.
Moreover individuals with an intersex condition may be more prone to psychological difficulties, due in part to behaviors of parents, relatives and other people.
DIAGNOSIS
Complete androgen insensitivity syndrome, as well as complete deficiency in testosterone synthesis, is not usually suspected in the affected individuals since they appear phenotypically as women.

Suspicion about the presence of this condition appears when the menses fail to develop at puberty, or an inguinal hernia presents during premenarche. From1 to 2% of girls that present an inguinal hernia will also have CAIS. A diagnosis of pseudohermaphroditism can be made in utero by comparing the karyotype obtained by amniocentesis with the external genitalia of the fetus during a prenatal ultrasound: the presence of a Y chromosome can be highlighted either by fluorescence obtained by in situ hybridization (FISH) or developing a full karyotype analysis.
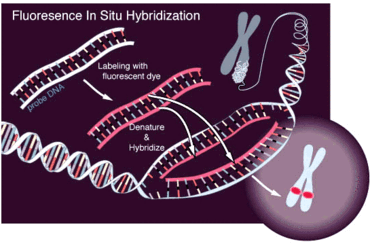
The FISH technique consists in hybridizing a fluorescent probe of nucleotides complementary in sequence to a short section of DNA on a target gene that in this case can be a gene set on Y chromosome. If the probe hybridizes to the target gene fluorescence can be visualized under a fluorescent microscope and it proves the presence of the researched gene.
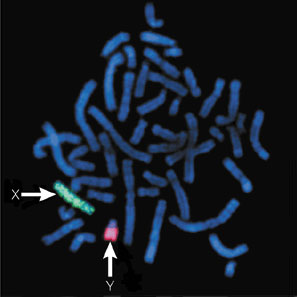
The diagnosis of CAIS is confirmed when AR gene sequencing reveals a mutation, although about 5% of individuals with CAIS do not have an AR mutation. At the same way, gene sequencing may be of fundamental importance to diagnose different mutations in genes encoding for 5-alpha-reductase, 17b HSD III and cytochrome P450c17.
It is current practice to disclose the karyotype at the time of diagnosis when the affected girl is at least of adolescent age. If the affected individual is a child or an infant, parents, together with a psychologist, have to decide when to disclose the diagnosis.
A diagnosis of partial androgen insensitivity syndrome or partial deficit of testosterone can be done at the moment of birth since these infants presents ambiguous genitalia that can appear in very different degrees of partial masculinization and feminization. However some tests have to be done to confirm the diagnosis in order not to confuse these syndromes with other pathological conditions which appears with similar phenotypes. One of the possibilities consist in checking for the presence of gene mutations through gene sequencing (even if AR gene mutations cannot be found in 27-72% of individuals with PAIS.) The human chorionic gonadotropin (hCG) stimulation test can be carried out in order to test whether the gonads are testes. If they are there will be an increase in the level of serum testosterone in response to the hCG stimulation, even if testes are not descended into the scrotum. An increased level of serum testosterone can help differentiate between androgen resistance/ lack and gonadal dysgenesis. Testicular function can also be tested by measuring serum anti-Müllerian hormone levels.
Mild androgen insensitivity syndrome is diagnosed in normal phenotypical males and is not typically researched except in cases of male infertility. In fact MAIS with its mild characteristics remains often unnoticed and untreated. It can be difficult to diagnosed even after semenological, clinical and laboratory data examination.
THERAPY
Since methods to correct a malfunctioning proteins that result from mutated gene are not currently available, management of affected individuals is limited to the treatment of symptoms.
Individuals with complete androgen resistance or lack are always raised as females and they do not have gender identity problems since their brain are not masculinized.
In patients affected by vaginal hypoplasia this pathological condition can be corrected using non-surgical pressure dilation methods. Dilation is possible even when the vaginal depth is significantly compromised thanks to the elastic nature of vaginal tissue. The non-surgical pressure dilation method is currently recommended as the first choice, since it is non-invasive and highly successful. When non-surgical pressure dilation methods fail and dilatation results impossible, the alternative solution is represented by surgical intervention.

Gonadectomy is highly recommended for individuals with cryptorchidism in order to mitigate gonadal tumor risk. If gonadectomy is performed early, puberty must be artificially induced administering gradually increasing doses of estrogen. On the other hand when gonadectomy is performed late, female puberty will occur on its own as a consequence of the aromatization of testosterone into estrogen. Women who are not compliant with estrogen replacement therapy, and have a subsequent loss of concentration of estrogen, may be affected by a significant loss of bone mineral density.
Progestin replacement therapy is seldom initiated because of its futile role due to the absence of a uterus.
It has also been proved that androgen replacement therapy may improve patients’ conditions, giving a sense of well-being in gonadectomized women. However the mechanism by which androgens can lead to this benefit is not entirely understood.
As regards psychological and social matters, parents of children affected by these syndromes often need a support in order to decide how and when disclose the diagnosis. The assistance of an expert psychologist is recommended both for parents and for children in order to help and face such a delicate situation.
The condition of ambiguous genitalia is related to another relevant and delicate problem that is sex assignment which corresponds with the decision of whether to raise an individual as a boy or a girl. Grades 3 and 4 of the Quigley and Prader scale in particular present phenotypes that cannot be classified neither as primarily male nor as a female. Arbitrary gender assignment should be avoided: it is recommended to wait for the affected individuals to decide for themselves. Decision depends on how the brain has developed during fetal life, that is due to how much testosterone has been able to masculinized sexual dimorphic areas of central nervous system.
In individuals with ambiguous genitalia, genitoplasty can be proposed as a solution. However since it can be irreversible and there is no guarantee that adult gender identity will develop as assigned by surgical intervention, it would be better delay the intervention and allow the affected patient to reach an age and maturity sufficient to have a role in such decision.
Mild androgen insensitivity syndrome often remains untreated because of its mild symptoms that are seldom recognized. However it has been proved that the administration of testosterone can improve secondary sexual characteristics typical of men with MAIS and it is also useful to reverse infertility due to this condition.
PROGNOSIS
People affected by pseudohermaphroditism often have to face psychological difficulties in accepting their condition and also difficulties with sexual function and infertility.
With appropriate medical and psychological treatment, individuals with complete female phenotypes can be satisfied with their sexual function and psychosexual development so that they can lead active and normal lives. At the same way, men with minimal feminization of genitalia incur in few problems that allow them to live a quite normal life. On the other hand patients with intermediate conditions may have many more difficulties since they present an ambiguous phenotypes and often an ambiguous sexual orientation that prevent them from leading a normal life and often also from having normal relationships with other people.
Pseudohermaphroditism, from Wikipedia, the free encyclopedia
Complete androgen insensitivity syndrome, from Wikipedia, the free encyclopedia
Partial androgen insensitivity syndrome, from Wikipedia,the free encyclopedia
Mild androgen insensitivity syndrome, from Wikipedia, the free encyclopedia
FEMALE PSEUDOHERMAPHRODITISM
PATIENT RISK FACTORS and PATHOGENESIS
Female pseudohermaphroditism is normally due to:
• Hyperandrogenism: that is an excess of androgens of extragonadal origin. The main cause of accumulation of androgens is Congenital virilizing adrenal hyperplasia
• Deficit in the synthesis of estrogens that eventually results in a similar accumulation of androgens
Accumulation of androgens in female fetus leads to partial masculinization of the external genitalia while internal organs are normally feminized as a result of the presence of XX karyotype.
Main cause of female pseudohermaphroditism:
Congenital virilizing adrenal hyperplasia
Congenital adrenal hyperplasia (CAH) is a form of adrenal insufficiency in which the enzyme that produces two important adrenal steroid hormones, cortisol and aldosterone, is deficient. Because cortisol and aldosterone production is impeded, the adrenal gland instead overproduces androgens (DHEA, androstenedione and testosterone.)
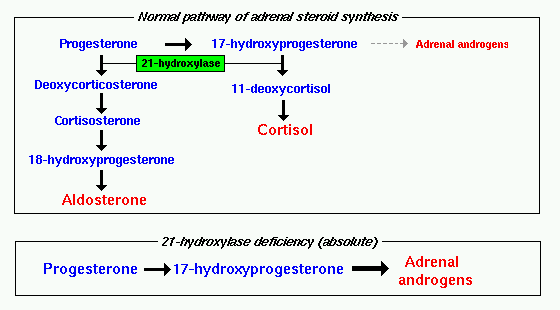
Congenital adrenal hyperplasia can be due to a deficient activity of the enzyme P450c21, also known as 21-hydroxylase (21-OH) and encoded by the gene CYP21A on chromosome 6p21.3. This enzyme is located in the smooth endoplasmic reticulum of the adrenal cortex cells. It catalyzes hydroxylation of 17-hydroxyprogesterone to 11-deoxycortisol in the glucocorticoid pathway leading to the production of cortisol. It also catalyzes hydroxylation of progesterone to 11-deoxycorticosterone in the mineralocorticoid pathway on its way from pregnenolone to aldosterone.
The gene for 21-hydroxylase has a pseudogene (CYP21P), 30 kb away from it, that is 98% homologous in structure to CYP21A. This pseudogene is inactive because of minor differences in the gene. The proximity of CYP21P with CYP21A is thought to predispose the CYP21A gene to crossovers in meiosis between CYP21A and CYP21P, resulting in loss of genetic function and subsequent loss of the enyme 21-hydrozylase. Other defects in this enzyme occur because of gene deletions or mutations. 95% of gene mutations are thought to be due to recombination with CYP21P, 20% are thought to represent deletions, and 70% are point mutations.
Deficient activity of this enzyme reduces serum cortisol levels: this condition leads to elevation of ACTH levels and subsequent hyperplasia of the adrenal gland. ACTH also stimulates uptake of cholesterol and synthesis of pregnenolone; as a result 17-hydroxypregnenolone and 17- hydroxyprogesterone serum level increases and it can reach from 10 to 1000 times the normal concentration. This accumulation of steroid precursors leads to an increased synthesis of DHEA, androstenedione, and testosterone.
It is important to notice that the deficit which causes this excess in the production of androgen can affect either fetus or mother’s adrenal gland. Both these conditions in fact cause an increment of fetal serum level of testosterone, responsible for the masculinization of external genitalia.
Other causes of congenital adrenal hyperplasia may involve the 11-beta-hydroxylase enzyme or the 3-beta-hydroxysteroid dehydrogenase which are both involved in the pathway that leads to synthesis of cortisol and androsterone.
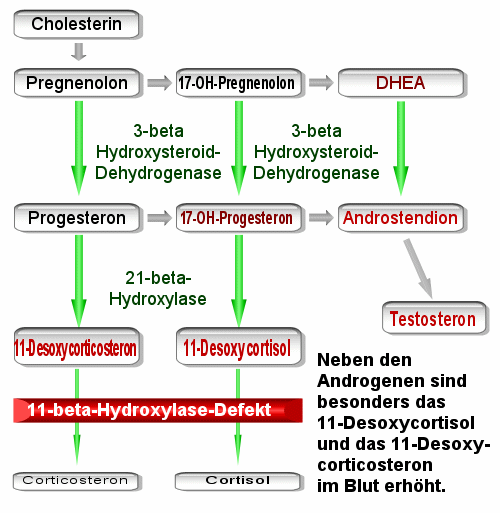
The 11-beta-hydroxylase gene is set on chromosomal band 8q21. The encoded enzyme catalyzes the conversion of 11-deoxycortisol to cortisol in the glucocorticoid pathway and the conversion of deoxycorticosterone to corticosterone in the mineralocorticoid pathway.
3-beta-hydroxysteroid dehydrogenase isoenzyme 1 is found in adrenal glands and gonads, while isoenzyme 2 is found primarily in the placenta and liver. The genes for both forms reside on chromosomal band 1p13. The classic form of 3-beta-hydroxysteroid dehydrogenase deficiency results from mutations or deletions in the gene for the adrenal form of the enzyme.
Both deficits of these enzyme result in a similar condition characterized, also in this case, by accumulation of steroid precursors and increased syntheses of androgens.
Pathophysiology of Congenital Adrenal Hyperplasia, from Medscape Reference. Author: Thomas A Wilson, MD; Chief Editor: Stephen Kemp
Unique steroid 21-hydroxylase gene CYP21A2 polymorphism in patients with hyperandrogenism signs
A unique case of female pseudohermaphroditism with 21-hydroxylase deficiency and small supernumerary marker chromosome 7. Al-Achkar W, Wafa A, Assaad M, Ehlers C, Liehr T.
Other genetic RISK FACTORS
Deficit in the synthesis of estrogens
Female pseudohermaphroditism can also be the result of a deficit of the aromatase enzyme, which catalyzes the conversion of androstenedione into estrone and the extremely important conversion of testosterone into estradiolo.
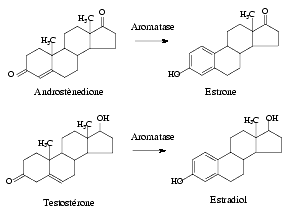
Aromatase is expressed in a wide range of tissues, including ovarian granulosa cells and luteal corpus, liver, muscles, breast, brain and testis.
P450 Aromatase CYP19 gene is set on chromosome 15q21.1. It is composed by 9 translated exons encoding a unique protein of 55 kDa and 11 untranslated intones at the beginning of the gene. Nineteen CYP19 gene mutations have been identified: four of them consist in substitutions in the coding exons; ten mutations are substitutions in the untranslated parts; six mutations are alterations in the 5′ untranslated region and one in the 3′ region. Point mutations are found predominantly in exons 9 and 10 and often leads to enzyme loss of funcion. Deficit of aromatase is an autosomic recessive disturb resulting in the impossibility of producing estrogens from androgen precursors and in the subsequent accumulation of androgens.
Aromatase Deficiency and Estrogen Resistance: From Molecular Genetics to Clinic, Departments of Obstetrics & Gynecology and Molecular Genetics, Division of Reproductive Endocrinology, University of Illinois at Chicago, Chicago, Illinois.
SYMPTOMS
Phenotypes and symptoms of female Pseudohermaphrodites

The presence of testosterone and other androgens during fetal live of female fetuses with a XX karyotype, obviously leads to genital ambiguity due to partial masculinization of external genitalia. The ovaries develop normally and, since they have not been exposed to testicular antimullerian hormone, the uterus, fallopian tubes, upper vagina, and other mullerian structures develop without problems.
On the other hand presence of high levels of testosterone results in masculinization of clitoris that can reach the size of a well developed phallus. In addition to this, the vaginal opening can be closed, as well as female urethra while a male urethra can appear inside the phallus. The labial skin may become as thin and rugated as a scrotum.
There are many degrees of masculinization external genitalia: the most severely affected female infants can even appear as a real male.

Prader scale (from stage 1 to stage 5) can also be used to classify female pseudohermaphrodites dependig on their phenotypes.
Stage 1 is characterized by a quite normal female phenotype with a quite enlarge clitoris that can be considered otherwise normal.
Stages 2 and 3 represent ambiguous intermediate phenotypes.
Stage 4 is characterized by a quite masculinized phenotype. However the phallus has a minimal size and the scrotum is empty, since these individuals have ovaries due to the XX karyotype.
Stage 5 correspond to a complete male phenotype. The penis has a quite normal size and the urethral opening is at the tip of it. The scrotum however continues to be empty. The vagina connects internally with the urethra.
Individuals affected by female pseudohermaphroditism often present a masculinization of sexual dimorphic areas of central nervous system, due to the presence of high level of testosterone during the fetal critical period of development. This condition can leads to severe problems of gender identity.
DIAGNOSIS
Individuals whose external genitalia are highly masculinized are supposed to be male affected by cryptorchidism. If they are affected by congenital adrenal hyperplasia, the diagnosis is not suspected until signs of salt-wasting develop a week later, due in particular to the lack of aldosterone.
When the external genitalia are ambiguous is necessary to investigate for the presence of a uterus and ovaries. The diagnosis can be confirmed when levels of serum testosterone result to be extensively increased while anti mullerian hormone is not present.
A full karyotype analysis can be useful to reveal the genotype XX of female pseudohermaphrodites infants; the CYP19 gene sequencing can reveal mutations responsible for the inactivation of aromatase while sequencing of genes encoding 21-hydroxylase, 11-beta-hydroxylase and 3-beta-hydroxysteroid dehydrogenase can reveal mutations in the corresponding enzymes.
THERAPY
In order to return to normal androgen, glucocorticoids and mineralocorticoids serum levels, in patients with congenital adrenal hyperplasia, hormone replacement therapy has to be administer during the all life.
One of the hormone replacement therapy role is to protect from adrenal insufficiency and to suppress the excessive adrenal androgen production. It is then of fundamental importance to replace glucocorticoids and mineralocorticoids in order to guarantee physiological functions and to suppress ACTH production.
Adrenalectomy can be consider a radical treatment for congenital adrenal hyperplasia and it is recommended for patients with little or no enzyme activity. Removal of adrenal glands has to be followed by hormone therapy.
Females who have masculine external genitalia require surgical intervention in order to reconstruct the clitoris and the vagina. However it may happen that female individuals prefer to maintain masculinized external genitalia since they have developed a male gender due to the masculinization of their brain. For this reason surgical reconstructive intervention is not recommended until the affected individuals can decide for themselves.
PROGNOSIS
People affected by pseudohermaphroditism may have relevant problem in accepting their condition.
However while individuals with a mild masculinization of the external genitalia as well as those who have complete male phenotype often do not present incompatibility of gender since they present feminized brain and high level of masculinization respectively, people having ambiguous genitalia and an ambiguous gender due to partial virilization of the brain may have serious problems resulting from the incompatibility of external aspect and male or female gender.
It is quite obvious that life for these individuals can be very difficult as well as relationships with other people.
Pseudohermaphroditism, from Wikipedia, the free encyclopedia
Congenital adrenal hyperplasia, From Wikipedia, the free encyclopedia
These syndromes should make us meditate on the fact that the contingency of an apparently minimal and insignificant mutation can deeply change ourselves, our lives and even our own way to see life through the eyes of a woman or the eyes of a man.
Silvia Mungo Gaspare Arrigo Sbirziola