DEFINITION
Proprotein convertase subtilisin/kexin type 9, also known as PCSK9, is an enzyme that in humans is encoded by the PCSK9 gene. PSCK9, is inactive when it is first synthesized, because it has a section of peptide chains that blocks its activity; proprotein convertases remove that section to activate the enzyme. PCSK9 has medical significance because it acts in cholesterol homeostasis. Drugs that block PCSK9 can lower low-density lipoprotein cholesterol (LDL-C).
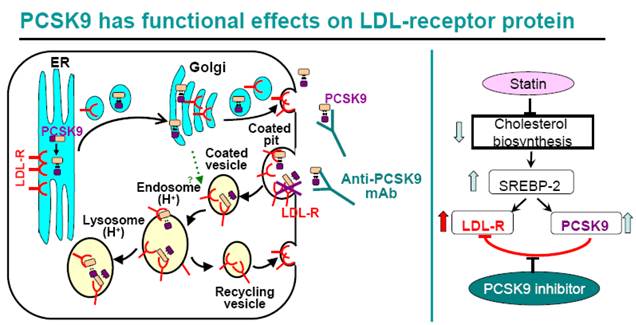
An Anti-PCSK9 Antibody Reduces LDL-Cholesterol On Top Of A Statin And Suppresses Hepatocyte SREBP-Regulated Genes , 2012
THE GENE
CHEMICAL STRUCTURE AND IMAGES
When relevant for the function
- Primary structure
- Secondary structure
- Tertiary structure
- Quaternary structure
Protein Aminoacids Percentage


The 2 proteins have many similarities (Chart 2) but also significant differences:
- LDLR high aspartate (high oxalacetate, high Krebs cycle rate?)
- PCSK9 high alanine low aspartate (high pyruvate?)
SYNTHESIS AND TURNOVER
mRNA synthesis
protein synthesis
post-translational modifications
Autocatalytic cleavage is required to transport it from the endoplasmic reticulum to the Golgi apparatus and for the secretion of the mature protein.
- Localizes to the endoplasmic reticulum in the absence of LDLR
- Colocalizes to the cell surface and to the endosomes/lysosomes in the presence of LDLR.
The sorting to the cell surface and endosomes is required in order to fully promote LDLR degradation.


In the rough endoplasmic reticulum (ER), the inactive form of proprotein convertase subtilisin kexin 9 (PCSK9) — the zymogen proPCSK9 (amino acids 31–692) —undergoes an intramolecular autocatalytic cleavage at its internal site (VFAQ152↓) to generate the prosegment (amino acids 31–152) and PCSK9 (amino acids 153–692) that remain tightly bound as a prosegment–PCSK9 complex28, 46. The enzymatically inactive tight binding complex of prosegment–PCSK9 exits the ER, traverses the Golgi and is constitutively secreted from the trans-Golgi network into the medium. If proPCSK9 cannot be processed (indicated by a red star), because of a heterozygous natural mutation (for example, Q152H) or owing to the presence of an inhibitor, it remains as a zymogen in the ER and binds the non-mutated wild-type sequence, thereby acting like a dominant negative mutant, and effectively preventing PCSK9 secretion. Upon reaching the trans-Golgi network, the wild-type prosegment–PCSK9 complex can either be sorted directly to lysosomes as a complex with the low-density lipoprotein receptor (LDLR) (intracellular pathway), or it is secreted and then internalized with the LDLR into clathrin-coated endosomes for lysosomal targeting and degradation, which requires the binding of the cytosolic adaptor protein autosomal recessive hypercholesterolaemia protein (ARH) (extracellular pathway). It is not yet clear whether the cell surface PCSK9 competes with apolipoprotein B (APOB) on the LDL particles for LDLR binding. In the absence of PCSK9, the endocytosed APOB–LDLR complex dissociates and the LDLR is recycled back to the cell surface. In the presence of PCSK9 and within the acidic milieu of endosomes the LDLR is not recycled, and (by a currently undefined mechanism) the PCSK9–LDLR tight binding complex is sent to lysosomes for degradation. (The biology and therapeutic targeting of the proprotein convertases, 2012)
degradation
CELLULAR FUNCTIONS
cellular localization,
biological function
- Cell signaling and Ligand transport
- Structural proteins
REGULATION
Statins treatment enhances PCSK9
DIAGNOSTIC USE
THERAPEUTIC USE
alirocumab (Sanofi, Regeneron) 3 fold more effective than Ezetimibe in reducing LDL-cholesterol