POMPE DISEASE
Epidemiology
The Pompe disease (glycogen storage disease type II or acid maltase deficiency) is a rare progressive autosomal recessive genetic disease, whose name is linked to the Dutch pathologist Johannes C. Pompe (1910-1945), who first described it.
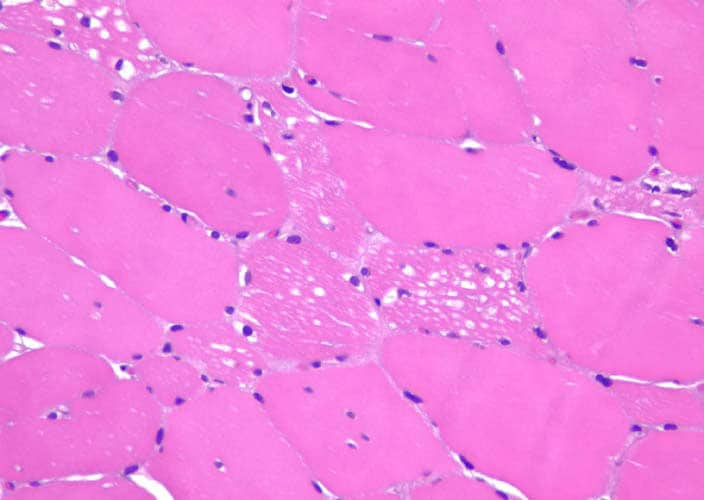
It is a metabolic myopathy characterized by generalized accumulation of glycogen inside lysosomes caused by deficiency of the lysosomal enzyme acid α glucosidase or acid maltase. This enzyme is needed to break down glycogen, a form of sugar stored in muscle cells. When muscle cells are deposited in an excessive amount of glycogen, the cells are damaged and the muscles are no longer able to function properly.
It has an estimated rate of 1/40.000 Afro-Americans, 1/50.000 Chinese, 1/146.000 Australian. The disease has long been considered untreatable, but in 2006 it’s been introduced the enzyme replacement therapy with recombinant human acid α-glucosidase that has been shown to alter the prognosis.
Etiology
This disease, progressive and often fatal, is caused by mutations in the gene located in 17q23 chromosome, which encodes the lysosomal enzyme acid α-glucosidase (GAA), which is essential for the degradation of glycogen to glucose. The total or partial deficiency of the enzyme activity of GAA results in the accumulation of glycogen within lysosomes, resulting in cell damage and tissue.
The multisystem disease is because, despite the accumulation of glycogen is more pronounced in muscle fibers, this is also observed in other tissues.
The presence of glycogen stores, which subvert the inside of the muscle fibercontractile apparatus, impairs muscle function significantly during contraction, with progressive degeneration of muscle fibers and fibroadipose replacement. The final consequence of this process is progressive muscle weakness, which can be observed in severe or advanced stages of the disease. However, other mechanisms appear to be involved in the pathogenesis of tissue damage. Studies have suggested in the 80’s, as a possible additional cause of loss of strength, excessive muscle proteolysis, secondary to the release of hydrolytic enzymes in the cytoplasm by the lysosomes, where it accumulates glycogen. Latest morphological studies have indicated in the autophagy, one of the mechanisms responsible for cellular toxicity by accumulation of glycogen, particularly in the fibers of the second kind. The excessive accumulation of intracellular autophagosome compromises the cell vesicular network and the function of muscle fiber. Finally, mitochondrial alterations, such as mitochondrial swelling and irregularities of cristae structure, are observed in the majority of muscle biopsies. Morphologically scattered or small groups of angular atrophic fibers are frequently seen in Pompe patients, a pattern which suggests neurogenic atrophy. These findings may be related to glycogen storage in anterior horn cells of the spinal cord, which may lead to motorneuron destruction.
Glycogen and lysosomes

Glycogen is the main reserve polysaccharide of animal cells. It is a polymer of glucose units joined with sub links (α1-> 4) and with branches that start with links (α1-> 6). Ramifications are every 8-12 residues and it’s present mainly in the liver but also in skeletal muscle. In cells is present in the form of large agglomerates or granules. When used as an energy source, glucose units are detached from the ends one at a time.
The degradation of glycogen occurs in different ways in lysosomes (1-3%) and in the cytoplasm.
Lysosomes take small amounts of glycogen with autophagy. They do not accumulate physiologically, because it is cleaved by an enzyme, but when this enzyme is not working then that glycogen accumulates to levels harmful because it is not degraded to glucose. Lysosomes are able to degrade glucose immediately thanks to the action of acid hydrolases in it, the alpha 1-4 glucosidase, which acts at the level of these organelles, with a pH of 4-5 (optimal for its function). This constitutes a minor pathway for the degradation of glycogen. The physiological regulation of this enzyme in due in to the muscle by epinephrine, AMP and calcium, in to the liver by glucagon and the concentration of glucose.
In the cytoplasm, however, is the major pathway, glycogen degradation involves the activities of phosphorylase (other than in the liver and muscle that needs to be converted to active form of action of a kinase). Also need the activity of an enzyme, called debrancher, able to move and separate branches of the oligosaccharides from the bonds 1-6.
The gene GAA
The official name of this gene is “glucosidase alpha acid.”

The gene spans approximately 20 kb and contains 20 exons with first exon being noncoding. The coding sequence of the catalytic site domain is interrupted in the middle by an intron of 101 bp. The GC content is high (80%) and distinct TATA and CCAAT motifs are lacking.
Most cases appear to be due to three mutations. A transversion (T → G) mutation is the most common among adults with this disorder. This mutation interrupts a site of RNA splicing.
Clinic
Pompe disease has a complex and heterogeneous clinical presentation. Depending on age of onset, three forms of the disease are recognized:
◗ the classical form and more severe, occurring shortly after birth and usually causes death within the first year of life;
◗ non-classical form, with onset between the first and second years of life, characterized by a variable prognosis;
◗ The late-onset form, which can occur at any age after the first year of life and is characterized by a slow progression and adverse outcomes less than the classic form.
The infantile-onset Pompe disease, the most aggressive form has as its most obvious manifestation of excessive weakness. Infants with Pompe disease are hypotonic and unable to lift his head. You may not be able to sit, to roll or crawl. The muscle weakness progresses rapidly. Breathing, sucking and swallowing become extremely difficult. The heart, liver and tongue swell. The growing weakness of the heart causes cardiac and respiratory failure. The development does not seem to be affected by mental illness.
The late-onset Pompe disease can appear in childhood or adulthood. The progressive muscle weakness and breathing problems are the main symptoms. The first symptom is often the weakness of the legs, causing gait unsteady or shaky. Patients may experience muscle pain and be subject to frequent falls. Older children can often be subject to the development of an abnormal curvature of the spine (lordosis, kyphosis or scoliosis) that persists into adulthood. Pompe disease affects the diaphragm. With the gradual weakening of the diaphragm, breathing becomes more difficult, especially during sleep. This leads to morning headaches and day time sleepiness. The children with Pompe disease may have trouble staying in the same way as their peers when performing physical activities. Adults can easily get tired and breathless after performing physical activities, or you climb the stairs. Some suffer from lower backpain. The enlargement of the heart or liver is rarely known in this late-onset form of the disease.
Musculoskeletal system
The damage is usually symmetrical and may involve all muscle groups, but affects the muscles more than proximal and distal lower limbs than the upper. The appearance of pseudo-hypertrophic often observed in some muscles, due to the accumulation of glycogen in the tissue. Muscle damage may also involve the muscles of mastication and tongue.
Respiratory system
Pompe disease is also associated to a considerable weakening of the respiratory muscles
(diaphragm, intercostal muscles and accessories). The involvement of respiratory muscles may occur early. The respiratory failure is the major cause of morbidity and mortality.
Central nervous system
The literature and clinical evidence that describes the signs and symptoms of neurological disease are very rare. Several post-mortem studies conducted in the early 70’s
have described the presence of glycogen stores and low GAA activity in various areas of the central nervous system. The deficiency of the enzyme activity was however without pathological or neurological symptoms.
Diagnosis
Pompe disease is not always easily recognizable. The variability and the wide spectrum of clinical phenotypes are partly due to different mutations in the GAA, which cause a complete or partial reduction of enzyme activity remaining. The measurement of the residual enzyme activity is decisive. After confirmation of the presence of a deficit in the activity of the GAA, it is advisable to characterize the mutation of the enzyme by performing molecular analysis.
Measurement of enzyme activity
There is a correlation between the level of residual activity and the severity of the disease.
The GAA activity was measured using a biochemical test in which you can use synthetic substrates (4-methylumbelliferyl-α-D-glucoside) or natural substrates (maltose, glycogen). In the classic forms of this disease is not possible to show the enzymatic activity but in later forms you can measure various levels of residual activity. The test can be performed on cultured fibroblasts, muscle biopsies, lymphocytes purified mononuclear cells, lymphoid cell lines.
Molecular analysis
After confirmation of the diagnosis by biochemical tests, identification of the mutationin the gene GAA is not strictly necessary. However, the molecular test is truly important because it allows us to identify conclusively the carrier of a mutation and is therefore strongly
recommended for all close relatives of a patient, especially if they want to have a child.
Histological exams
In case of marked muscle symptoms, muscle biopsy can help diagnose. Muscle biopsies can also be used to measure the amount of glycogen stored.
Imaging
More useful methods are CT and MRI. Compared to CT, MRI is more sensitive in identifying the replacement fibroadipose and has higher resolution.
Symptoms control
The assessment of muscle strength can be performed by several validated tests. The most common is the manual muscle testing, which measures the strength of different muscle groups quantified by the scale-points .
The pulmonary function tests (spirometry, pulmonary volumes) should be performed together with assessment of respiratory muscle strength.
Therapy
The disease has long been considered untreatable, but in 2006 was introduced the enzyme replacement therapy with recombinant human acid α-glucosidase (Myozyme), that has been shown to alter the prognosis. The commonly used dose is 20 mg/kg administered by intravenous infusion once every 14 days.
It’s been shown that a diet enriched with protein, promotes protein synthesis in the muscle. The aerobic exercise can help reducing the storage of glycogen and increasing protein synthesis in muscles. Physiotherapy is useful to prevent contractures in order to correct posture and any deformities.
Bibliography
Web
• http://ghr.nlm.nih.gov/gene/GAA
• http://www.uniprot.org/uniprot/P10253
• http://harvester.fzk.de/harvester/human/IPI00293/IPI00293088.htm
• http://en.wikipedia.org/wiki/Acid_alpha-glucosidase
• http://www.wkhealth.it/_upload/oaj/8_Pompe.pdf
• www.ncbi.nlm.nih.gov/pmc/articles/.../pdf/biochemj00512-0015.pdf
• dispense
• http://amdapompe.ehclients.com/downloads/publications/Therapeutic_Approaches.pdf
• http://it.wikipedia.org/wiki/Morbo_di_Pompe
• http://www.orpha.net/consor/cgi-bin/OC_Exp.php?lng=IT&Expert=365
• www.associazionecmn.com/.../Glicogenosi_tipo_II-Malattia_di_Pompe.pdf
• http://www.genzyme.it/aree-therapeutiche/malattia-di-pompe.aspx
• www.wkhealth.it/_upload/oaj/8_Pompe.pdf
Pompe's disease 2009":http://www.ncbi.nlm.nih.gov/pubmed/18929906