Francesca Maria Elena Frigiolini, Pietro Giacopelli

DEFINITION
Classic galactosemia, also called galactosemia type 1, galactose-1-phosphate uridylyltransferase deficiency or GALT deficiency, is an autosomal recessive. disorder , caused by an inborn deficency of enzyme galactose-1-phosphate uridylyltransferase ( GALT ) involved in the metabolism of galactose.
The conseguent accumulation of galactose in the blood is a poison to the body and can cause serious complication.
Classic galactosemia belongs to the family of Galactosemias . Researchers have identified several types of galactosemia.
These conditions are each caused by mutations in a particular gene, and affect different enzymes involved in breaking down galactose:
- Classic galactosemia is the most common and most severe form of the condition. It is caused by the deficiency of galactose-1-phosphate-uridylyl transferase;
- Galactosemia type II is caused by the deficiency of galactokinase;
- Galactosemia type III is caused by the deficiency of UDP-Galactose-4-epimerasi
STORY
The first case in literature of the condition that we know to be galactosemia was reported by Von Reuss, in 1908 (Von reuss, A., Zuckeratisseheidung in Sdiuglingsalter, WVien med.Wchschr.).
His work was basically post mortem. Von Reuss described the case of an infant who began to lose weight at eight weeks of age, though it had developed normally up to that time.
When seen by Von Reuss, the child weighed 2% pounds less than his birth weight.
Examination revealed enlargement of both liver and spleen. The patient had a definite galactosuria.
The galactosuria ceased when his diet was changed from one simulating breast milk to one of tea and dextrose or to a cereal made with flour instead of milk.
Von Reuss felt that continuation of the diet was not indicated because of the infant's poor condition.
Actually, had he continued the dietary treatment the patient might have lived. The infant died after three weeks' hospitalization with the clinical diagnosis of cirrhosis of the liver.
At postmortem examination the liver was markedly enlarged and the surface finely granular; the histological findings confirmed the diagnosis of cirrhosis.
However, the first detailed description of classic galactosemia was given by Goppert in 1917 (Goppert, F. Galaktosurie nach Milchzuckergabe bei angeborenem, familiaerem chronischem Leberleiden. Klin. Wschr. 54: 473-477, 1917.).
The proband (A.G.) presented with large liver, icterus, failure to thrive, and urinary excretion of albumen and sugar.
After exclusion of galactose from the diet, these signs and symptoms normalized.
He was mentally retarded (developmental quotient of 14 months at 36 months of age). He tolerated sucrose, maltose, glucose, and fructose at doses of 2 g/kg, but after lactose or galactose there was dose-dependent galactosuria.
His oldest brother had suffered from icterus and liver enlargement a few days after birth and had had a life-threatening bleed after ritual circumcision. He died after 6 weeks.
At autopsy, a huge liver tumor was present, and the cause of his death was attributed to nephritis.
His third sib, born somewhat prematurely, became icteric, and died after 4 weeks.
Goppert concluded that the patient was suffering from a familial liver disorder and that in such cases lactose must be replaced by another sugar.
The causes of classical galactosemia were identifided only some years later, in 1956, by a group led by Herman Kalckar
For more details see the article by W.A.Wilson .
EPIDEMIOLOGY
The incidence of classic galactosemia is about 1 per 30 000 live births for Caucasians.
In other populations the incidence rate differs. For example, galactose-1-phosphate uridylyltransferase deficiency is relatively common among the Travellers (1 per 480), an endogamous group of commercial/industrial nomads within the Irish population, while the disorder is thought to be much less common in "Asians": http://www.ncbi.nlm.nih.gov/pubmed/21150919 .
Galactosemia equally affects males and females. Galactosemia is most often diagnosed in infancy by newborn screening because all states include galactosemia as part of their newborn screen.
Aside from the high mortality rate in newborn infants with sepsis caused by Escherichia Coli , life expectancy has never been studied in patients with galactosemia.
Informations from Medscape
GENETICS
| Location | Phenotype | Phenotype MIM number | Gene/Locus | Gene/Locus MIM number |
| 9p13,3 | Galactosemia | 230400 | GALT | 606999 |
The gene for galactose-1-phosphate uridylyltransferase ( GALT gene ) is located on the short arm of chromosome 9, in the region 9p13.
The gene is approximately 4 kb in length and has 11 exons and 10 introns. The promoter is GC rich as in a housekeeping gene.
Significant genotype-phenotype correlations have been noted .
Today, More than 130 mutations in the GALT gene have been identified in people with the classic form of galactosemia.
No other phenotypes are associated with GALT mutations. Most of these mutations severely reduce or eliminate the activity of galactose-1-phosphate uridylyltransferase .
A shortage of this enzyme prevents cells from processing galactose obtained from the diet.
Most changes in the GALT gene alter single protein building blocks (amino acids) used to build galactose-1-phosphate uridylyltransferase.
The most common GALT mutation in Caucasian Europeans and North Americans replaces the amino acid glutamine with the amino acid arginine at position 188 in the enzyme (written as Gln188Arg).
Another mutation occurs almost exclusively in people of African descent. This genetic change substitutes the amino acid leucine for the amino acid serine at position 135 (written as Ser135Leu).
A particular GALT mutation called the Duarte variant results in a form of galactosemia with less serious complications than the classic type.
This mutation replaces the amino acid asparagine with the amino acid aspartic acid at protein position 314 (written as Asn314Asp).
The Duarte variant reduces but does not eliminate the activity of galactose-1-phosphate uridylyltransferase.
The signs and symptoms associated with this variant tend to be milder because the enzyme retains 5 percent to 20 percent of its normal activity.
HEREDITABILITY
Classic galactosemia is inherited in an autosomic recessive manner , meaning parents of proband must have at least one mutation in the GALT gene.
- Homozygotes for the classic galactosemia (G) allele (G/G) have GALT enzyme activity less than 5% of control values;
- Heterozygotes for the classic galactosemia allele and a normal (N) allele(G/N) have GALT enzyme activity of approximately 50% of control values. They are called carriers and have no symptoms.
Each child from two carrier parents would have a 25% chance of being affected, a 50% chance of being a carrier, and a 25% chance of inheriting normal versions of the gene from each parent.

BIOLOGY AND PHYSIOLOGICAL FUNCTION OF GALACTOSE-1-PHOSPHATE URIDYLYLTRANSFERASE
Galactose-1-phosphate uridylyltransferase ( GALT ) is an enzyme responsible for converting ingested galactose to glucose. This enzyme has been shown to belong to the histidine triad (HIT) super family .
Members of this family function as either nucleotide hydrolases or transferases that act upon the a-phosphorus of nucleotides.
Galactose-1-phosphate uridylyltransferase ( GALT ) catalyzes the second step of the Leloir pathway of galactose metabolism.
Galactose is a C-4 epimer of glucose. It is found in dairy products, sugar beets, and other gums and mucilages. It is also synthesized by the body, where it forms part of glycolipids and glycoprotein in several tissues. The main pathway of galactose metabolism is the Leloir pathway. It consists of the latter stage of a two-part process that converts ß-D-galactose to the more metabolically useful UDP-glucose .
In the initial stage ß-D-galactose is epimerized to a-D-galactose by galactose mutarotase ( GALM ).
The Leloir pathway then carries out the conversion of a-D-galactose to UDP-glucose through three principle enzymes:
- Galactokinase ( GALK ): catalyzes the ATP-dependent phosphorylation of a-D-galactose to yield galactose 1-phosphate;
- Galactose-1-phosphate uridylyltransferase ( GALT ): catalyzes the reversible transfer of a UMP group from UDP-glucose to galactose 1-phosphate , thereby generating glucose 1-phosphate and UDP-galactose . Phosphoglucomutase 1 (PGMU) then catalyzes formation of the Glucose-6-phosphate from Glucose 1-phosphate. Hydrolysis of Glucose 6-phosphate to Glucose is catalyzed by Glucose-6-phosphatase, catalytic subunit (G6PT).
- UDP-galactose 4-epimerase (GALE): converts UDP-galactose to UDP-glucose. UDP-glucose participates in formation of Glycogen catalyzed by two enzymes, glycogen synthase 2 (liver) (GYS2) and by glycogen synthase 1 (muscle) (GYS1). Glycogen is the substrate of one more reaction of formation Glucose 1-phosphate catalyzed by Glycogen phosphorylase.

Of the enzymes in the Leloir pathway, only the reaction catalyzed by galactose-1-phosphate uridylyltransferase proceeds through a covalently bound intermediate. According to the proposed mechanism, UDP-glucose binds to the enzyme, a uridylylated enzyme intermediate is generated, and glucose 1-phosphate is released.
Subsequently, galactose 1-phosphate binds to the active site and the UMP moiety is transferred to generate UDP-galactose.
In the enzyme from E. coli, His-166 has been shown to be the residue transiently modified
Additional information on galactose metabolism
CHEMICAL STRUCTURE

The three-dimensional structure of the enzyme from E. coli was elucidated by Wedekind et al. in 1995 .
The enzyme is a homodimer. Each subunit of the enzyme contains 348 amino acid residues and additionally one zinc and one iron.
The overall fold of the subunit has been referred to as a “half-barrel” with nine strands of anti-parallel ß-sheet flanked on either side by a-helices.
The iron serves in a structural capacity by bridging two ß-strands and an a-helix near the subunit: subunit interface of the dimer. The zinc ion is located within ~8 Å of the active site and is tetrahedrally ligated by Cys-52, Cys-55, His-115, and His-164. On the basis of amino acid sequence alignments, it appears that in higher organisms Cys-52 and His-115 are not conserved.
In the figure above, the two subunits of the dimeric enzyme are displayed in red and blue with the positions of the metals indicated by theround spheres. The active site in this protein model contains bound UDP-glucose.
To trap the uridylyl-enzyme intermediate, single crystals of the active enzyme were transferred to solutions containing UDP-glucose and moved to successively higher pH values up to 7.1.
Under these conditions, the enzyme was active, but the rate of acid-catalyzed hydrolysis of the intermediate was reduced.
This study revealed a covalent bond between N of His-166 and the a-phosphorus of UMP. Additionally it was shown that the side chain of Gln-188 provided important hydrogen bonds to both O2 and O5' of the nucleotide. In the human enzyme, the mutation of this glutamine to an arginine is the predominant cause of galactosemia among the Caucasian population.
This change to an arginine residue may result in overstabilization of the enzyme intermediate, thereby compromising its subsequent reaction with galactose 1-phosphate.
Studies revealed that the active site for the uridylyltransferase is formed by amino acid residues contributed by both subunits of the homodimer. Accommodation of the glucose versus galactose moieties is accomplished by simple movements of two side chains and by a change in the backbone dihedral angles of Val-314.
PATHOGENESIS
The pathogenesis of classic galactosemia is still not well understood. It is attractive to assume that the clinical manifestations are the direct result of accumulation of galactose within a range of cell types and tissues, leading to disturbed cell/organ function, but this explanation is likely to be simplicistic.
In classic galactosemia, galactose-1-phosphate uridylyltransferase activity is reduced or absent, leading to an accumulation of galactose and Gal-1-P.

Galactose can undergo two destinies:
- Reduction to galactitol : in galactosemic patients, the accumulation of galactose becomes the substrate for enzymes that catalyze the polyol pathway of carbohydrate metabolism. The first reaction of this pathway is the reduction of aldoses, types of sugars including galactose, to sugar alcohols. Recent data suggests that aldose reductase is the enzyme responsible for the primary stage of this pathway.
Therefore aldose reductase reduces galactose to its sugar alcohol form, galactitol. Galactitol, however, is not a suitable substrate for the next enzyme in the polyol pathway, polyol dehydrogenase. Thus, galactitol accumulates in body tissues and is excreted in the urine of galactosemic patients. Accumulation of galactitol has been attributed to galactosemic cataract, and high concentrations of galactitol have been found in people with classic galactosemia;
- Oxidation to galactonate : the mechanism of galactonate formation is still unclear. However, recent studies suggest that galactose dehydrogenase is responsible for converting galactose to galactonolactone, which then spontaneously or enzymatically converts to galactonate. Once formed, galactonate may enter the pentose phosphate pathway . Thus, oxidation to galactonate serves as an alternate form of metabolizing galactose. This oxidative pathway renders accumulated galactonate less harmful than accumulated galactitol.
Although above average levels of galactitol and galactonate were observed in patients with galactosemia, is not yet understood whether and how they can cause damage to the body. High level of galactitol and palatinate were observed in
Instead it seems that the liver lesions are not caused by galactitol and galactonate, but by a still unknown metabolic product .
Informations from Insights into the pathogenesis of galactosemia
GALACTOSEMIC CATARACT PATHOGENESIS
Different discourse give rise to the pathogenesis of galactosemic cataract.
The mechanism by which galactosemia causes cataract is not well understood, but the topic has been approached by researchers for decades. Through this collective effort, a general mechanism for galactosemia’s causation of presenile cataract has come into form.
In galactosemic cataracts, osmotic swelling of the lens epithelial cells occurs. Researchers concluded that this osmotic swelling must be the result of an accumulation of abnormal metabolites or electrolytes in the lens. Ruth Van Heyningen was the first to discover that the lens’s retention of galactitol, induces this osmotic swelling in the galactosemic cataract.
However, galactose concentration must be fairly high before the enzyme, aldose reductase, will convert significant amounts of the sugar to its galactitol form.
As it turns out, the lens is a favorable site for galactose accumulation. The lens phosphorylates galactose at a relatively slow pace in comparison to other tissues. This factor, in combination with the low activity of galactose-metabolizing enzymes in galactosemic patients, allows for the accumulation of galactose in the lens.
Aldose reductase is able to dip into this galactose reservoir and synthesize significant amounts of galactitol.
The sugar alcohol idly begins to accumulate in the lens. As galactitol concentration increases in the lens, a hypertonic environment is created.
Osmosis favors the movement of water into the lens fibers to reduce the high osmolarity. This osmotic movement ultimately results in the swelling of lens fibers until they rupture.
Vacuoles appear where a significant amount of osmotic dissolution of fiber has taken place. What are left are interfibrillar clefts filled with precipitated proteins: the manifestation of a cataract.
Friedenwald was able to show that periphery lens fibers always dissolve before fibers at the equatorial region of the lens. This observation has been confirmed by more recent experiments as well, but is still unexplained.
The progression of galactosemic cataract is generally divided into three stages; initial vacuolar, late vacuolar, and nuclear cataract.
The formation of a mature, nuclear, cloudy galactosemic cataract typically surfaces 14 to 15 days after the onset of the galactose diet.
Although advancement has been slow to come during the decades of research dedicated to the galactosemic cataract, some notable additions have been made.
Essentially, the study suggested that the mechanism which centers on osmotic swelling of the lens fibers, is just the beginning in a cascade of events that causes and progresses the galactosemic cataract. Mulhern determined that osmotic swelling is actually a cataractogenic stressor that leads to LEC apoptosis.
This is because osmotic swelling of lens fibers considerably strains LEC endoplasmic reticula. As the endoplasmatic reticulum is the principal site of protein synthesis, stressors on the ER can cause proteins to become misfolded.
The subsequent accumulation of misfolded proteins in the ER activates the unfolded protein response (UPR) in LECs. UPR initiates apoptosis by various mechanisms, one of which is the release of ROS .
Thus, according to recent findings, osmotic swelling, UPR, oxidative damage, and the resultant LEC apoptosis all play key roles in the onset and progression of the galactosemic cataract.
Other studies claim that the oxidative damage in LECs is less a result of the release of ROS and more because of the competition between aldose reductase and glutathione reductas for NADPH.
Aldose reductase requires NADPH for the reduction of galactose to galactitol, while glutathione reductase utilizes NADPH to reduce glutathione disulfide to its sulfhydryl form, GSH. GSH is an important cellular antioxidant. Therefore, what exactly the key roles are for these cataractogenic factors is not yet fully understood or agreed upon by researchers.
More information on galactosemic cataract
SYMPTOMS AND COMPLICATIONS
| Finding | Percent | Additional Details |
| Hepatocellular damage | 89% | Jaundice, Hepatomegaly, Abnormal liver function test, Coagulation disorders, Ascites |
| Food intollerance | 76% | Vomiting, Diarrhea, Poor feeding |
| Failure to thrive | 29% | Hepatocellular damage |
| Lethargy | 16% | - |
| Seizures | 1% | - |
| Sepsis | 10% | Escherichia Coli, Klebsiella, Enterobacter, Staphylococcus, Beta-Streptococcus, Streptococcus Fecalis |
Infants with classic galactosemia have no GALT enzyme activity and are unable to convert galactose to glucose. Initial clinical symptoms of galactosemia, which usually begins within a few days of ingesting breast milk or lactose-containing formulas, are:
- Feeding problems: baby refuses to eat formula containing milk;
- Hepatocellular damage and especially Jaundice of intrinsic liver disease: macroscopically, the initial pathologic changes are characterized by enlargement of the organ which has smooth and yellow or greenish-yellow.
With the passing of time, the liver becomes more hard, fibrous, with characters similar to those of cirrhosis.
Histologically, at the beginning, is observed in the liver fatty degeneration or vacuole, which results in a framework of hepatocyte necrosis.
The regressive degenerative fenoma stimulates hepatocyte proliferation, so after a while, you can see the processes of regeneration of hepatocytes, with architecture that recalls the lobular structure. Together with the regressive degenerative processes, occurring phenomena of colangiolar proliferation periportal and phenomena of connective proliferation reparative consequent to hepatocyte necrosis.
The colangiols contain bile thrombi and are surrounded by inflammatory infiltrates granulocytic neutrophils.
Finished process of hepatocyte proliferation, in the liver parenchyma can be observed outbreaks erythrocyte, and hepatocytes and deposits of pigment emosiderinico in the interstices. Then the typical changes of cirrhosis appears.
Information from Lezioni di anatomia patologica
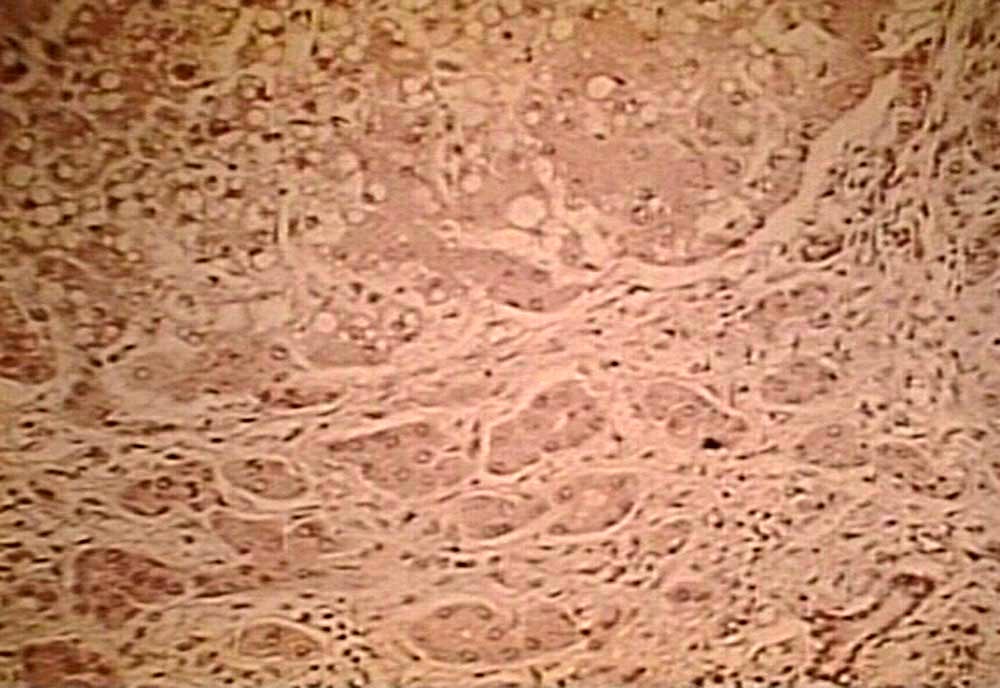
If a lactose-/galactose-restricted diet is provided during the first three to ten days of life, the symptoms resolve quickly and prognosis is good. If the diagnosis of galactosemia is not established, the infant who is partially treated with intravenous antibiotics and self-restricted lactose intake demonstrates relapsing and episodic jaundice and bleeding from altered hemostasis concomitant with the introduction of lactose.
If classic galactosemia is not treated or treatment is delayed, complications and long term out-come may occur. The following details on long-term outcome were reported by Waggoner et al [1990] as the result of a retrospective, cross-sectional survey of 270 individuals with classic galactosemia:
- Oil-drop-cataract : caused by accumulation of galactitol within the lens cells leads to increased intracellular osmotic pressure and fluid influx into the lens.Early cataracts reversible with dietary restriction of galactose. Zonular or nuclear opacity with increased refractive power of the nuclear portion causes an "oil droplet" appearance on retroillumination. This type of cataract may be found only on slit-lamp examination and missed with an ophthalmoscope, since it consist of punctate lesions in the fetal lens nucleus . Galactosemic cataracts were reported in 30% of 314 individuals. Nearly half the cataracts were described as "mild," "transient," or "neonatal" and resolved with dietary treatment; only eight were treated surgically. Dietary treatment had begun at a mean age of 77 days for those with cataracts compared to 20 days for those without cataracts. However, one of the eight individuals who required cataract surgery was an infant who had been treated from birth. See also

- Cirrhosis of the liver ;
- Kidney damage;
- Delayed speech development: difficulty speaking correctly and consistently. Problems were reported in 56% of individuals age three years or older. More than 90% of the individuals with speech problems were described as having delayed vocabulary and articulation problems, also called verbal dyspraxia . The speech problem resolved in only 24%. A recent, more formal analysis found speech problems in 44% of individuals; 38% had a specific diagnosis of developmental verbal dyspraxia [Robertson & Singh 2000, Webb et al 2003]. The developmental quotients and IQ scores observed in individuals with speech disorders as a group were significantly lower than those of individuals with normal speech; however, some individuals with speech problems tested in the average range;
- Mental retardation mental retardation is a condition diagnosed before age 18 that includes below-average general intellectual function, and a lack of the skills necessary for daily living. In the brain of affected people are found decrease in the number of the nerve cells, phenomena of gliosis and edema, especially in correspondence of the cerebral cortex, of the olives or of the dentate nuclei of the cerebellum. Probably these lesions are not characteristic of galactosemia, but are the result of hypoglycemia, always present in affected individuals. Mental retardation is attributed to these brain injuries.
Of 177 individuals who were at least age six years and had no obvious medical causes for developmental delay other than galactosemia, 45% were described as developmentally delayed. The mean IQ scores of the individuals as a group declined slightly (4-7 points) with increasing age. Studies of Dutch individuals at various ages using a quality of life questionnaire indicated subnormal cognitive outcomes;
- Motor function: among individuals older than age five years, 18% had fine-motor tremors and problems with coordination, gait, and balance. Severe ataxia was observed in two teenagers;
- Sepsis with Escherichia Coli ;
- Premature ovarian insufficiency (POI): is the loss of function of the ovaries before age 40. Of 47 girls and women, 81% had signs of POI. POI may be manifest as cutaneous rashes in estrogen-depleted children. The mean age at menarche was 14 years with a range from ten to 18 years. Eight out of 34 women over age 17 years (including two with "streak gonads") had primary amenorrhea. Most women developed oligomenorrhea and secondary amenorrhea within a few years of menarche. Only five out of 17 women over age 22 years had normal menstruation. Two, who gave birth at age 18 and 26 years, had never experienced normal menstrual periods. Pregnancy is rare in women with galactosemia because of the high frequency of hypergonadotropic hypogonadism with ovarian atrophy. Normal serum concentrations of testosterone and/or follicle-stimulating hormone (FSH) and luteinizing hormone (LH) were reported for males;
- Failure to thrive: in many individuals, growth was severely delayed during childhood and early adolescence; when puberty was delayed and growth continued through the late teens, final adult heights were within the normal range. Decreased height over mean parental height was related to decreased IGF-I ;
- Death.

Infants who survive the neonatal period and who continue to drink milk that contains galactose develop intellectual disability and other cortical and cerebellar tract signs.
Even with early and adequate therapy, the long-term outcome in older children and adults with classic (G/G) galactosemia can include cataracts, speech defects, poor growth, poor intellectual function, neurologic deficits (predominantly extrapyramidal findings with ataxia), and premature ovarian insufficiency (POI). Outcome can be predicted based on the level of GALT enzyme activity, GALT genotype, age at which successful therapeutic control was achieved, and compliance with lactose restrictions.
Daniel’s story
DIAGNOSIS
Clinical Diagnosis
Most affected infants are detected through newborn screening programs; however, clinicians need to be alert to early signs (poor feeding, prolonged neonatal jaundice) and remove lactose from the diet and initiate soy-based, dietary therapy while awaiting results of newborn screening and/or diagnostic tests.
Testing
- Biochemical assays : necessary for diagnosis and monitoring of therapy include the following:
- Erythrocyte galactose-1-phosphate concentration.
Metabolism of this precursor is blocked in the GALT reaction sequence. Concentration of erythrocyte galactose-1-phosphate exceeds 2 mg/dL and can be used to monitor the effectiveness of therapy.
In classic galactosemia, gal-1-P remains elevated between 2 and 5 mg/dL despite therapy.
- Galactitol. A product of an alternate pathway for galactose metabolism, galactitol can be measured in the urine. Urinary galactitol greater than 78 mmol/mol creatinine is abnormal.
- Total body oxidation of 13C galactose to 13CO2 . Elimination in breath of less than 5% of 13C galactose as 13CO2 two hours after administration of 13C-D galactose defines a severe metabolite phenotype. Such testing is used in Phase II research protocols and may become useful as an early screen for galactosemia before discharge from the nursery.
- GC/MS isotope dilution method . Experimental measurements of galactitol and galactonate in urine are made by the GC/MS isotope dilution method .
- Measure of bone mineral density : diminished bone mineral density (BMD) is a well known complication in women with classic galactosaemia caused by premature ovarian failure.
- Proton magnetic resonance spectroscopy: brain edema may occur in infants with galactosemia.
NewBorn Screening

Galactosemia can be detected in virtually 100% of affested infants in states that include testing for galactosemia in their newborn screening programs ( National Newborn Screening Status Report )
- Newborn screening utilizes a small amount of blood obtained from a heel prick to:
- assay galactose-1-phosphate uridyltransferase (GALT) enzyme activity;
- quantify total red blood cell (RBC) gal-1-P concentration and galactose.
Newborn screening tests are most commonly done from whole blood samples collected on specially designed filter paper. The filter paper is often attached to a form containing required information about the infant and parents.
This includes date and time of birth, date and time of sample collection, the infant's weight and gestational age.
The form will also have information about whether the baby has had a blood transfusion and any additional nutrition the baby may have received.
Ideally, newborn screening samples are collected from the infant between 24 hours and 7 days after birth Samples are mailed daily to the laboratory responsible for testing. Most jurisdictions require samples to be collected for screening from all newborns, unless the parent or guardian opts out of the process in writing.
The goal is to report the results within a short period of time. If screens are normal, a paper report is sent to the submitting hospital and parents rarely hear about it.
- If an abnormality occurs, employees of the agency, usually nurses, begin to try to reach the physician, hospital, and/or nursery by telephone.
They are persistent until they can arrange an evaluation of the infant by an appropriate specialist physician. The specialist will attempt to confirm the diagnosis by repeating the tests by a different method or laboratory, or by performing a second tier of molecular testing for specific GALT mutations which is the most sensitive and specific laboratory test.
The confirmatory test varies depending on the initial screen, and can include enzyme assays, DNA testing, Gas Chromatography/Mass Spectrometry.
Depending on the likelihood of the diagnosis, the specialist will initiate treatment and provide information to the family. Performance of the program is reviewed regularly and strenuous efforts are made to maintain a system that catches every infant with these diagnoses.
More information on newborn screening .
Molecular Genetic Testing:
- Targeted mutation analysis: mutation analysis for the eight common GALT galactosemia mutations (Gln188Arg, Ser135Leu, Lys285Asn, Leu195Pro, Tyr209Cys, Phe171Ser, 5kdel, c253-2A>G) is available on a clinical basis. The 5kbdel allele is a complex deletion that involves a 3163-nt deletion of the GALT promoter and a 5‘ gene region along with a 2295-bp deletion of the 3‘ gene (). The 5kdel complex deletion is detectable by any of a variety of methods that detect deletions (Southern blot analysis, PCR,…);
- Sequence analysis: identifies the common mutations identified by targeted mutation analysis as well sequence variants such as small intragenic deletions/insertions, missense, nonsense, and splice site mutations;
- Deletion/Duplication analysis: deletion of GALT exons and multi-exons has been detected in affected individuals.
More information on genetic testing
Prenatal Testing
Prenatal testing is possible for fetuses at 25% risk for classic (G/G) galactosemia using either GALT enzyme activity or molecular genetic testing if the two disease-causing GALT mutations in the family are "known":"http://www.ncbi.nlm.nih.gov/pubmed/11288121" .
Analysis of GALT enzyme activity and molecular diagnosis rely on cells obtained by chorionic villus sampling (CVS) at approximately ten to 12 weeks' gestation or amniocentesis usually performed at approximately 15 to 18 weeks' gestation. When a fetus has GALT deficiency, amniotic fluid concentration of galactitol is elevated in the late third trimester.
Prenatal testing of a treatable condition may be controversial if the testing is being considered for the purpose of pregnancy termination rather than early diagnosis.
Differences in perspective may exist among medical professionals and in families regarding the use of prenatal testing. Although most centers would consider this to be the choice of the parents, discussion of these issues is appropriate.


Infectious diseases, obstructive biliary disease including Alagille syndrome, progressive familial intrahepatic homeostasis and citrin deficiency and other metabolic diseases including Niemann-Pick disease type C and Wilson disease are in the differential diagnosis for neonatal hepatotoxicity.
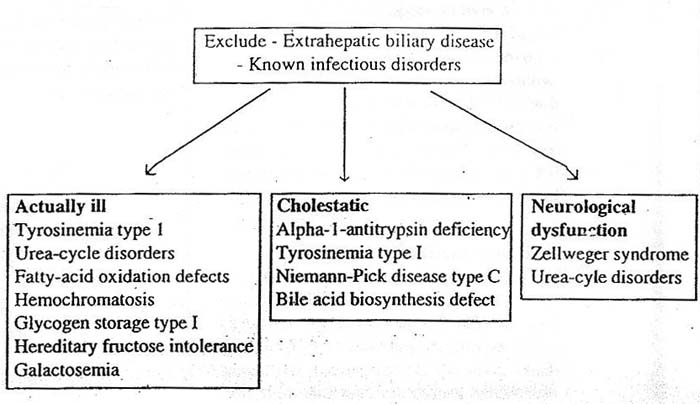
PATIENT RISK FACTORS
The primary risk factor for classic galactosemia is having family members with the disease or who are carriers of the disease. In particular:
- Parents of a proband: the parents of an affected individual are obligate heterozygotes and therefore carry one mutant allele;
- At conception, each sib of a proband with G/G galactosemia has a 25% chance of being affected, a 50% chance of being a carrier of a disease-causing allele, and a 25% chance of having two normal alleles;
- Once an at-risk sib is know to be unaffected, the chance of his/her being a carrier is 2/3.
- affected females are at increased risk for premature ovarian insufficiency, but may have children;
- Offspring of one parent with (G/G) galactosemia and one parent with two normal alleles (N/N) are obligate heterozygotes (G/N);
- If one parent is affected (G/G) and the other parent is a carrier for a G allele (G/N), each child has a 50% chance of being a heterozygote and a 50% chance of having G/G galactosemia.
- Other family members of a proband: each sib of the proband's parents is at a 50% risk of being a carrier.
It is appropriate to offer genetic consueling (including discussion of potential risks to offspring and reproductive options) to young adults who are affected or at risk. The optimal time for determination of genetic risk, clarification of carrier status, and discussion of the availability of prenatal testing is before pregnancy.
TREATMENT
Treatment Of Manifestation
- Lactose restriction reverses liver disease in newborns who already have hepatocellular disease;
- POI: stimulation with FSH may be useful in producing ovulation in some women. Others have found that POI in classic galactosemia may be treatable by exogenous pharmacologic stimulation by gonadotropic hormones. See.. .
- Galactosemic cataract: the treatment for galactosemic cataract is no different from general galactosemia treatment. Actually, galactosemic cataract is one of the symptoms reversible. Infants should be immediately removed from a galactose diet when symptoms present, and the cataract should disappear and visibility should return to "normal. See.. .
- Developmental evaluation, and focus on speech development with appropriate interventions are recommended;
- Failure to thrive: today is not curable.
Prevention Of Primary Manifestation
- Dietary intervention: immediate dietary intervention is indicated in infants whose GALT enzyme activity is less than 10% of control activity and whose RBC galactose-1-phosphate is greater than 10 mg/dL. Because 90% of the newborn’s carbohydrate source is lactose and human milk contains 6%-8% lactose, cows' milk 3%-4% lactose, and most proprietary infant formulas 7% lactose, all of these milk products must be replaced immediately by a formula that is free of bioavailable lactose (e.g., Isomil® or Prosobee®).
Such soy formulas contain sucrose, fructose, and non-galactose polycarbohydrates.
Continued treatment with soy-based formula depends on the response of elevated erythrocyte gal-1-P: concentrations lower than 5 mg/dL are considered within the therapeutic range.
Dietary restrictions on all lactose-containing foods (dairy products, tomato sauces, and candies) and medicines (tablets, capsules, sweetened elixirs that contain lactulose) should continue throughout life; however, managing the diet becomes less important after infancy and early childhood, when milk and dairy products are no longer the primary source of energy.
It is debated how stringent the diet should be after the first year of life, as endogenous galactose production is an order of magnitude higher than that ingested from foods other than milk.


Prevention Of Secondary Complication
Therapies Under Investigation
Research suggests that despite exogenous galactose restriction, endogenous galactose production may approach 2.0 g/day . If this is true, "self-intoxication" with galactose may be more of a problem than restriction of galactose from exogenous sources in the management of older children and adults who no longer depend on milk as their primary source of energy. Approaches to lowering endogenous production of gal-1-P are under investigation using small inhibitors of the GALK enzyme. Although in vitro studies of GALT enzyme-deficient human fibroblasts demonstrated proof of concept, a GALT enzyme-deficient mouse model is needed that expresses an ARHI signal. Note that GALT knockout mice do not express the human phenotype of galactosemia and have lost ARHI (DIRAS3) during evolution.
For more detailed information"
LINK