DEFINITION
Menkes disease is a X-linked, multisystemic disorder of copper metabolism. Affected patients are unable to adequately absorb and use copper, a micronutrient required for energy metabolism, catecholamine biosynthesis and connective tissue formation. In the classic form of MD patients have severe neurodegeneration and develop little or no motor and language skills; they also manifest kinky, hypopigmented hair and tortuous blood vessels.
Severely affected patients usually die in early childhood (before the third year of life); though variable forms exist: occipital horn syndrome (OHS) is the mildest one.
As it is inherited as an X-linked recessive trait, the majority of patients are males. MD is due to mutations in the ATP7A gene and most are intragenic mutations or partial gene deletions. ATP7A is an energy dependent transmembrane protein involved in the delivery of copper to the secreted copper enzymes and in the export of surplus copper from cells.
Biochemical markers of the disease include low serum levels of copper and caeruloplasmin and altered cerebrospinal fluid catechol levels.
A cure for the disease does not exist, but very early copper-histidine treatment may correct some of the neurological symptoms.
EPIDEMIOLOGY
The incidence of the disease is estimated to range between 1:40 000 and 1:350 000. Approximately 25% of the ATP7A mutations are gross deletions ranging in size from a single exon to the whole gene except for the first two exons.
About half of the point mutations (deletions/insertions/duplications and nonsense mutations) are truncating mutations, which result in a non-functional truncated protein. One interesting aspect is that in the copper-binding domains of ATP7A no missense mutations are observed. This suggests that variations in this region are more acceptable and do not necessarily lead to MD. In contrast, missense mutations in the domains important in catalysis (A- and P-domains) are poorly tolerated.
Females affected
Female carriers are mosaics of wild-type and mutant cells due to the random X inactivation, and they are rarely affected. The clinical symptoms of affected females are generally milder than those of affected boys with the same mutations.
Females affected http://www.ncbi.nlm.nih.gov/pmc/articles/PMC3298521/?tool=pubmed
SYMPTOMS
Complete loss‐of‐function mutations in ATP7A result in MD, whereas less severe defects produce the allelic disorder occipital horn syndrome (OHS).
Classical MD
The most common severe form of MD is characterized by progressive neurodegeneration and connective tissue dysfunction.
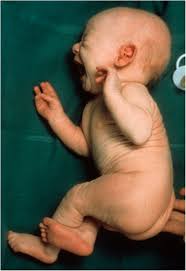
Pregnancy is usually uncomplicated.
Then, though, it is possible to observe:
* cephalohematomas and spontaneous fractures at birth
* prolonged jaundice in the early neonatal period
* hypothermia
* hypoglycemia and feeding difficulties
* pectus excavatum and umbilical and inguinal hernias
Sparse and lusterless scalp hair at the age of 1–2 months, together with pale skin and a rather expressionless appearance may be the first sign. Initial psychomotor development is usually unremarkable. Up to about 2–4 months of age the baby ceases to develop further and gradually loses some of the previously developed skills. The developmental regression becomes obvious around 5–6 months of age. Additional symptoms are failure to thrive, poor eating, vomiting, and diarrhea.
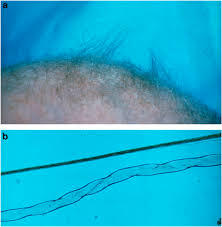
Spasticity and weakness of the extremities cause spontaneous movements to become limited and drowsiness and lethargy to emerge. The patients are typically diagnosed at 3–6 months of age, often due to the abnormal hypopigmented or depigmented hair, that resembles like steel wool; it is lusterless and friable, especially in the areas of the scalp subjected to friction.
Vascular, urogenital, and skeletal abnormalities are common. Skeletal changes like pectus excavatum or pectus carrinatum, and generalized osteoporosis bring on spontaneous fractures. The joints are hyperextensive, and loose and dry skin may be observed very early. Seborrheic dermatitis is also a frequent feature. In later stages patients frequently fail to follow visual stimulus.
Late manifestations are blindness, subdural hematoma, and respiratory failure. Most patients die within the third year of life due to infection, vascular complications or the neurological degeneration itself.
The OHS
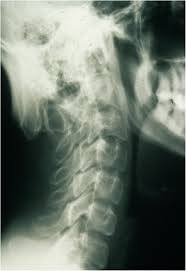
OHS is the mildest form of MD, and its principal clinical features are related to connective tissue. The main distinction between OHS and the other forms of MD is the radiographic observation of characteristic occipital horns. These are symmetric exostoses protruding from the occipital bone and pointing down.
Pregnancy is normal. Within days, hypothermia, jaundice, hypotonia, and feeding problems may develop. The first signs that bring the child to medical attention may be frequent diarrhea or urinary tract infections. Diagnosis is usually made only around 5–10 years of age.
Motor development is delayed due to muscular hypotonia, height is usually normal, while mild disproportion with long trunk, narrow chest and shoulders, thoracolumbar kyphosis or scoliosis, and pectus deformity are common. The joints are hyper mobile. Facial appearance gradually becomes distinctive: long, thin face, often with a high forehead, down-slanting eyes, hooked or prominent nose, long philtrum, high arched palate, and prominent large ears. Hair is not conspicuously abnormal, although some patients may have lusterless hair. Recurrent inguinal hernia is common together with vascular anomalies, such as varicose veins and arterial aneurysms. A particular problem is orthostatic hypotension. The intellectual capacity is described as low to borderline normal.
The clinical course is characterized by chronic diarrhea, bladder diverticulae with recurrent urinary tract infections and occasional spontaneous bladder ruptures, orthostatic syncope, and joint instability in the inferior extremities. Life expectancy in OHS is variable, but longer than that in MD.
DIAGNOSIS
Initial diagnosis of MD is suggested by clinical features (especially typical hair changes) and supported by demonstration of reduced levels of serum copper and ceruloplasmin. In this period, plasma catecholamine analysis indicative of dopamine β-hydroxylase deficiency may be the choice as a rapid diagnostic test.
Other useful laboratory investigations are arteriography, computed tomography, magnetic resonance imaging, and radiography.
Radiographs of patients with classical MD show specific abnormalities: generalized osteoporosis, metaphyseal flaring and spurs in the long bones, diaphyseal periosteal reaction and thickening, and Wormian bones in the cranial sutures. Rib fracture due to osteoporosis is a common finding. In OHS patients occipital horns are characteristic radiographic findings. They continue to grow up to early adulthood.
Light microscopy of hair shows individual hairs that are twisted about their own axes (pili torti), with varying shaft diameters (monilethrix), and fragmentation at regular intervals (trichorrhexis nodosa).
A definitive biochemical test for MD exists and is based on intracellular accumulation of copper due to impaired efflux. Accumulation is evaluated in cultured fibroblasts, by measuring radioactive copper (64Cu) retention after a 20-h pulse, and impaired efflux is directly determined after a 24-h pulse–chase period. The ultimate diagnostic proof of MD is the demonstration of the molecular defect in ATP7A.
Prenatal diagnosis
In at-risk families only male fetuses need to be evaluated, and rapid sex determination can be made using Y-chromosome-specific DNA sequences.
Carrier determination by measuring radioactive copper accumulation in cultured fibroblasts is not reliable due to random X-inactivation, and mutation analysis should be performed.
In pregnancies, in the first trimester the total copper content in chorionic villi can be measured directly by sensitive and accurate methods like neutron activation analysis and atomic absorption, and in the second trimester copper accumulation is measured in cultured amniotic fluid cells.
PATHOGENESIS
Copper is an essential trace element in the body, since it is required for normal function of several copper enzymes involved in cellular respiration (cytochrome-c oxidase); neurotransmitter biosynthesis (dopamine β-hydroxylase); maturation of peptide hormones (peptidyl α-amidating enzyme); free-radical scavenging (superoxide dismutase); melanin production (tyrosinase); and iron homeostasis (ceruloplasmin and hephaestin). Copper has further been implicated in myelination, in regulation of the circadian rhythm and may also be necessary for coagulation and angiogenesis.
Cu transporters CTR1, ATP7A, and ATP7B ensure that adequate Cu is available for Cu-requiring processes and prevent excess Cu accumulation within cells. Copper can exist in two oxidation states (Cu1+ and Cu2+) and reversible interchange between these two states is the basis of the enzymatic reactions. The same property, however, can result in the production of free radicals, being so highly toxic. Fine regulation of copper homeostasis is, therefore, vitally important.
Copper metabolism in the cell
Copper uptake across the plasma membrane uses CTR1, an energy-independent membrane transporter. Metallothionein, glutathione and copper-specific chaperones are instead proteins that, binding copper in the cytoplasma, protect cell from the toxic effects of the free ion and guide it to different cellular locations, securing efficient delivery of the metal to the enzymes.
In the trans-Golgi network (TGN) two homologous membrane-bound, copper-specific ATPases, ATP7A and ATP7B (defective in MD and Wilson disease, respectively), transfer copper across the membrane into the lumen of the TGN, where it is delivered to secreted enzymes. ATP7A is expressed in almost every organ except the liver, where ATP7B is predominantly expressed. This explains why MD is a systemic disorder, while in Wilson disease mainly the liver is affected.
ATP7A and B have a dual role in the cell: apart from copper-loading of enzymes in the secretory pathway, they are responsible for ATP-driven efflux of copper from the cells. Under normal physiological copper concentrations ATP7A is localized to TGN, transporting copper into the lumen to the copper-dependent enzymes. Under increased copper concentrations ATP7A is translocated to the vesicles or to the plasma membrane.
Whole body copper metabolism
The dietary copper absorbed is about 1 mg/day, from the intestinal lumen across the mucosal barrier into the interstitial fluid, and to portal blood. The non-specific metal transporters, DMT1, ATP7A, and CTR1, are involved in this multistage process.
From the blood the copper is transported to the liver and in lesser amounts to the kidney and other tissues such as brain. In the liver, copper is either secreted to the blood bound to ceruloplasmin or excreted to the bile. Both processes are controlled by ATP7B; defective ATP7B will, thus, lead to increased amounts of copper in the liver and other organs as observed in Wilson disease patients. The main excretory route of copper is bile, while urinary loss is far less important.
Free copper ions are virtually non-existing in living organisms, where the metal is mainly bound to ceruloplasmin, albumin, and histidine. Copper is transported to the brain across the blood–brain barrier at the cerebral endothelium and the blood–cerebrospinal fluid barrier at the choroid plexus because CTR1, ATP7A, and ATP7B are all expressed a lot in brain barrier fractions.
Copper homeostasis in MD
The defect in Menkes disease is the reduced transport of dietary Cu across the basolateral membrane of enterocytes to hepatic portal circulation. This results in the entrapment of Cu within the intestinal mucosa and Cu deficiency in the blood and peripheral organs. Cu deficiency is further compounded by a reduction in Cu transport across the blood-brain barrier into the central nervous system.
In the liver of MD patients the low copper amount is instead due to requirement of the metal in other tissues, rather than defective metabolism, as in the normal liver ATP7B is the main copper transporter and not ATP7A.
The reason for the low copper content in the brain of MD patients is however more complex. Mammalian brain is one of the richest copper-containing organs in the body. Regulation of brain copper level is not well understood, but ATP7A must play a role, since MD leads to low copper levels in the brain. In MD patients copper is likely trapped in both the blood–brain barrier and the blood–cerebrospinal fluid barrier, while the neurons and glial cells are deprived of copper.
Neuronal demyelination is also observed in MD patients due to ATP7A inactivation.
Copper metabolism http://www.ncbi.nlm.nih.gov/pmc/articles/PMC2987322/?tool=pubmed#bib1
THERAPY
Careful medical care, and possibly copper administration, may extend life span up to 13 years or even more. A number of severely affected MD patients with long survival have been reported.
The objective of a specific treatment for MD is to provide extra copper to the tissues and copper-dependent enzymes. Oral administration of copper is ineffective as copper is trapped in the intestines; it should be supplemented parenterally or subcutaneously. Among the available copper compounds, copper histidine has proved to be the most effective, but it depends on early initiation and presence of at least partially functional ATP7A.
Copper–histidine treatment, however, could not prevent skeletal abnormalities and cannot be accepted as a definitive cure for MD, despite some reported successful outcomes.
BIBLIOGRAPHY
http://www.ncbi.nlm.nih.gov/pmc/articles/PMC2987322/?tool=pubmed#bib1
http://www.ncbi.nlm.nih.gov/pmc/articles/PMC3065767/?tool=pubmed :look for other correlation regarding role of ATP7A such as: glutamatergic signaling in neurons, macrophage bactericidal activity, roles in Alzheimer’s disease, role in vascular smooth muscle cells
http://www.ncbi.nlm.nih.gov/pmc/articles/PMC3298521/?tool=pubmed