TRANSFUSION-RELATED ACUTE LUNG INJURY
DESCRIPTION
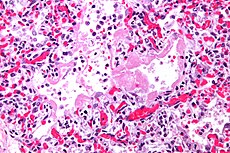
In medicine, transfusion related acute lung injury (TRALI) is a serious blood transfusion complication characterized by the acute onset of non-cardiogenic pulmonary edema following transfusion of blood products. It Is typically associated with plasma components such as platelets and Fresh Frozen Plasma, though cases have been reported with packed red blood cells since there is some residual plasma in the packed cells. Transfusion-related acute lung injury (TRALI) is an uncommon syndrome that is due to the presence of leukocyte antibodies in transfused plasma. Leukoagglutination and pooling of granulocytes in the recipient's lungs may occur, with release of the contents of leukocyte granules, and resulting injury to cellular membranes, endothelial surfaces, and potentially to lung parenchyma.
(Platelets induce NET in TRALI,2012)
CLINICAL PRESENTATION
Symptoms of TRALI typically develop during, or within 6 hours of a transfusion. Patients present with the rapid onset of dyspnea and tachypnea. There may be associated fever, cyanosis, and hypotension. Clinical exam reveals respiratory distress and pulmonary crackles may be present with no signs of congestive heart failure or volume overload. Chest x-ray (CXR )shows evidence of bilateral pulmonary edema unassociated with heart failure (non-cardiogenic pulmonary edema), with bilateral patchy infiltrates, which may rapidly progress to complete "white out" indistinguishable from Acute Respiratory Distress Syndrome (ARDS).

RISK FACTORS
The risk factors associated with TRALI are sepsis, aspiration of gastric contents, and multiple transfusion. Other risk factors where the incidence of TRALI is less clear include cirrhosis, toxic inhalation, pneumonia, neurogenic pulmonary edema, pancreatitis, prolonged hypotension, trauma, and lung resection. In addition, there may be yet other unidentified risk factors. Comorbid factors include older age, chronic alcoholism, tobacco abuse, absence of diabetes, greater severity of illness, and variant surfactant gene in women. Although multiple unit transfusion has been identified as a risk, transfusion of a small number of units was not studied as a risk factor for TRALI.
INCIDENCE
Incidence has not been well established due to difficulty in defining the syndrome and to variable reporting mechanisms worldwide. Various studies have estimated the overall frequency of TRALI to be between 1/1,120 and 1/57,810 units transfused. However, there is wide discrepancy in the literature with the reported frequency is as low as 1/557,000 RBC units and as high as 1/432 platelet units. TRALI is associated with a high morbidity with the majority of patients requiring ventilatory support. However, the lung injury is generally transient with PO2 levels returning to pretransfusion levels within 48 -96 hours and CXR returning to normal within 96 hours. TRALI is associated with a significant mortality rate, often approximated at 5 to 10%. Given the gains in safety made within the blood component production industry, particularly with respect to transmission of infectious diseases, TRALI is now among the three leading causes of transfusion related fatalities. (TRALI,2009)
PATHOGENESIS
TRALI is most often associated with transfusion of whole blood, packed red blood cell (pRBCs) and fresh frozen plasma (FFP). There are also reports of TRALI following transfusion of granulocytes cryoprecipitate platelet concentrates and plateletpheresis . It is hypothesized that TRALI may be precipitated by the infusion of donor antibodies directed against recipient leukocytes. The infusion of donor anti-HLA (human leukocyte antigens) or anti-HNA (human neutrophil antigens) antibodies is thought to directly cause complement activation, resulting in the influx of neutrophils into the lung, followed by neutrophil activation and release of neutrophil extracellular net (NET) that contains cytotoxic element with subsequent endothelial damage and capillary leak. Donor derived antibodies to HLA class I antigens and neutrophils have been demonstrated in up to 89% of TRALI cases examined in the literature. An alternate hypothesis argues that TRALI is the result of at least two independent clinical events: the first is related to the clinical condition of the patient (infection, cytokine administration, recent surgery, or massive transfusion) that causes activation of the pulmonary endothelium. This then leads to the sequestration of primed neutrophils to the activated pulmonary endothelium. The second event is the infusion of donor derived anti-HLA or anti-HNA antibodies directed against antigens on the neutrophil surface and/or biological response modifiers in the stored blood component that activate these adherent, functionally hyperactive neutrophils, causing neutrophil-mediated endothelial damage.

The most important role in the onset of TRALI seems to be due to neutrophil activation that induces the pathological phenotype by releasing neutrophil extracellular trap.
DISTRIBUTION OF NEUTROPHILS
In a healthy adult, 36% of all neutrophil reside in the circulating pool and 64% reside in a non-circulating ‘reserve’ pool, in the spleen and the bone marrow .If one compares the number of neutrophil in different anatomical locations, 28% of all PMNs are in the “pulmonary pool”; Importantly, the number of PMNs in the pulmonary pool does not stay constant, but increases in systemic inflammatory conditions such as TRALI.

PHENOTYPES of NEUTROPHILS
There are three functional phenotypes of neutrophils: quiescent, primed and activated.

- Quiescent phenotype:In healthy individuals the circulating neutrophils are usually in this state, they are free flowing in the circulation, have an approximate round morphology, are not adhesive, and have minimal membrane ruffling. They are recruited to the activated endothelium via released chemokines and are tethered, and captured by various selectins.
- Primed Phenotype: When neutrophils change from being non-adherent to being firmly adherent to they are considered to be primed.
However, primed neutrophils are functionally hyperactive but they do not induce release of theyr intracellular components.
They become larger and less deformable thus increasing their pulmonary transit times , especially in inflammatory states and in injured tissue.
- Activated phenotype: activated neutrophils are able to release NET.
(The Role of Neutrophils in the Pathogenesis of TRALI,2009)
NEUTROPHIL EXTRACELLULAR TRAPS (NET)
These structures are composed of DNA in association with histones, as the most abundant proteins in NET, as well as granular proteins such as elastase and myeloperoxidase and several cytoplasmic proteins. Inflammatory stimuli such as interleukin-8, lipopolysaccharide (LPS) or phorbol myristate acetate (PMA) provoke “NETosis” of neutrophils. During this cell-destructive process, which is distinct from classical apoptosis or necrosis, intracellular organelle membranes disintegrate after decondensation of materials in the nucleus, allowing the mixing of cytoplasmic and nuclear components which is followed by the rupture of plasma membrane to expel NET. These structures can bind and kill bacteria and fungi ,whereby NET-associated proteins such as elastase and histones exhibit bactericidal activity .
However, excessive activation of neutrophils may lead to the development of multiple organ dysfunction syndrome, it was also found that NETS trigger platelet and neutrophil deposition in the lungs, thus thrombosis and disseminated intravascular coagulation
(NET Directly Induce Epithelial and Endothelial Cell Death,2012)
There are three important component of NET:
Elastase is able to mediate neutrophil-induced tissue damage and efficiently
degrades extracellular matrix components .
Neutrophil elastase (or leukocyte elastase) also known as ELA2 is a serine proteinase of the same family as chymotrypsin and has broad substrate specificity. Secreted by neutrophils and macrophages during inflammation destroys bacteria and causes tissue damage. As with other serine proteinases it contains catalytic triad of histidine, aspartate, and serine. The gene encoding neutrophil elastase, ELA2, consists of five exons. It is one of the two human forms of elastase. The neutrophil form of elastase is 218 amino acids long, with two asparagine-linked carbohydrate chains.It is present in azurophil granules in the neutrophil cytoplasm.
Function: Elastases form a subfamily of serine proteases that hydrolyze many proteins in addition to elastin. Humans have six genes that encode the structurally similar proteins elastase 1, 2, 2A, 2B, 3A, and 3B. Elastase 2 hydrolyzes proteins within specialized neutrophil lysosomes, called azurophil granules, as well as proteins of the extracellular matrix following the protein's release from activated neutrophils. Elastase 2 may play a role in degenerative and inflammatory diseases by its proteolysis of collagen –IV and elastin of the extracellular matrix.

MPO have a significant role in defence against bacteria by conversion of hydrogen peroxidase to hypochlorous acid. Nevertheless, MPO activity can also provoke damage to the adjacent tissues; therefore, it may contribute to the pathogenesis of several inflammatory diseases including pulmonary injury such as TRALI. It has been reported that MPO can provoke caspase-3 activation and apoptosis . Moreover, MPO can induce DNA strand breakage in lung epithelial cells and so contribute to the cell-damaging capacity of NET.
(Inside the Neutrophil Phagosome,1998)
In biology, histones are highly alkaline proteins found in eukaryotic cell nuclei that package and order the DNA into structural units called nucleosomes. They are the chief protein components of chromatin, acting as spools around which DNA winds, and play a role in gene regulation. Without histones, the unwound DNA in chromosomes would be very long.
Five major families of histones exist: H1/H5, H2A, H2B, H3, and H4. Histones H2A, H2B, H3 and H4 are known as the core histones, while histones H1 and H5are known as the linker histones.

Despite their physiologic nuclear localization, nucleosomes have been found in the circulation of both healthy subjects and patients, where they can be released from dying cells or actively secreted by activated inflammatory cells (neutrophils, basophils, and mast cells) in the form of “extracellular traps,” complex structures of DNA strands, histones, and cell-specific granule proteins. High blood levels of nucleosomes have been detected in several inflammatory disease such as TRALI.
Recently has been found that histones particularly H4, can activate platelets through TLR2- and TLR4-dependent mechanisms.
It has been demonstrated that histones presented in neutrophil extracellular traps induce a pro coagulant phenotype in human platelets, thereby accelerating blood coagulation and enhancing thrombin generation, and that TLR2 and TLR4 are involved in the platelet response to histones.
(Extracellular histones promote thrombin generation through platelet-dependent mechanisms,2011)
PLATELET ACTIVATION
Platelets are widely recognized as mediators of inflammation and immune responses. They have been shown to express the TLRs, a major class of pattern-recognition receptors involved in the innate immune response through the recognition of microbial structures conserved among species (pathogen-associated molecular patterns) and of endogenous molecules released from damaged cells (damage-associated molecular patterns). TLR1, TLR2, TLR4, TLR6, TLR8, and TLR9 have been found on human platelets and were shown to be functional. For example, a TLR2 agonist induces platelet adhesion, degranulation, aggregation, and the formation of platelet-neutrophil aggregates and TLR4 agonist, induces platelet P-selectin expression and ATP secretion and primes platelets to aggregate in response to low-dose thrombin. Therefore, platelet TLRs might represent a bridge between inflammation and coagulation.
TOLL-LIKE RECEPTOR (TLR)
Toll-like receptors (TLRs) are a class of proteins that play a key role in the innate immune system They are single, membrane-spanning, non-catalytic receptors usually expressed in sentinel cells such as macrophages and dendritic cells, that recognize structurally conserved molecules derived from microbes.
Signaling:
TLRs are believed to function as dimers. Heterodimers or homodimer TLRs may also depend on other co-receptors for full ligand sensitivity.
A set of endosomal TLRs comprising TLR3, TLR7, TLR8 and TLR9 recognize nucleic acid derived from viruses as well as endogenous nucleic acids in context of pathogenic events. Activation of these receptor leads to production of inflammatory cytokines as well as type I interferons.
Four adapter molecules are known to be involved in signaling.
TLR signaling is divided into two distinct signaling pathways, the MyD88-dependent and TRIF-dependent pathway.

The MyD88-dependent response occurs on dimerization of the TLR receptor, Its primary effect is activation of NFκB and Mitogen-activated protein kinase. Ligand binding and conformational change that occurs in the receptor recruits the adaptor protein MyD88, a member of the TIR family. MyD88 then recruits IRAK 4, IRAK1 and IRAK2. IRAK kinases then phosphorylate and activate the protein TRAF6, which in turn polyubiquinates the protein TAK1, as well as itself in order to facilitate binding to IKKβ. On binding, TAK1 phosphorylates IKKβ, which then phosphorylates IκB causing its degradation and allowing NFκB to diffuse into the cell nucleus and activate transcription and consequent induction of inflammatory cytokines.
TRIF activates the kinases TBK1 and RIP1, which creates a branch in the signaling pathway. The TRIF/TBK1 signaling complex phosphorylates IRF3 allowing its translocation into the nucleus and production of Interferon type I. Meanwhile, activation of RIP1 causes the polyubiquination and activation of TAK1 and NFκB transcription in the same manner as the MyD88-dependent pathway.
TLRs pathways are one of the most important signals that promote gene modulation
and in this case promote the activation of the COAGULATION CASCADE.
In presence of histones, Platelet activation mediated by tlr induces the release of polyphosphate by dense granules, Polyphosphate are linear anionic polymer, and they have a powerful pro-coagulant activity that increases the coagulation capacity of 20 times . PolyP accumulates in a variety of microorganisms but they can also be secreted by activated human platelets; The two type of PolyP have different length (the one secreted by platelet are shorter than those released by microorganisms ), but both are potent modulator of the human blood clotting system, acting at 3 points in the clotting cascade:
- PolyP promote the contact pathway of blood clotting in an FXII-dependent manner (requiring very long PolyP polymers for optimal activity) accelerates FXI activation by α-thrombin, β-thrombin, and FXIa : polyP polymers of the size secreted by activated human platelets are very active in stimulating FXI activation by thrombin.
- PolyP Accelerate the activation of factor V by thrombin and factor Xa : PolyP secreted by human platelets and shorter PolyP have the same ability to promote this factor.
- PolyP enhance the thickness of fibrin fibrils: shorter polyP polymers, of the size secreted by activated human platelets, were far less potent than long-chain PolyP.
(Polyphosphate is a cofactor for the activation of factor XI by thrombin,2011)
COAGULATION CASCADE
Coagulation (thrombogenesis) is the process by which blood forms clots. It is an important part of hemostasis, the cessation of blood loss from a damaged vessel, wherein a damaged blood vessel wall is covered by a platelet and fibrin-containing clot to stop bleeding and begin repair of the damaged vessel. Disorders of coagulation can lead to an increased risk of bleeding (hemorrhage) or obstructive clotting (thrombosis).
The coagulation cascade has two pathways which lead to fibrin formation. These are the contact activation pathway (also known as the intrinsic pathway), and the tissue factor pathway (also known as the extrinsic pathway).
the pathways are a series of reactions, in which a zymogen (inactive enzyme precursor) of a serine protease and its glycoprotein co-factor are activated to become active components that then catalyze the next reaction in the cascade, ultimately resulting in cross-linked fibrin.
The coagulation factors are generally serine proteases (enzymes), which act by cleaving downstream proteins. The coagulation factors circulate as inactive zymogens. The coagulation cascade is classically divided into three pathways. The tissue factor and contact activation pathways both activate the "final common pathway" of factor X, thrombin and fibrin.
Long and short chain PolyP stimulate the activation of FXI so the Intrinsic pathways is involved:

The contact activation pathway or intrinsic pathways begins with formation of the primary complex on Factor XIa (the letter a indicate the active form of the factor) activates FIX, which with its co-factor FVIIIa form the tenase complex, which activates FX to FXa.
Here start the common pathways in which FXa and co-factor convert prothrombin to thrombin which is primary role is the conversion of fibrinogen to fibrin, the building block of a hemostatic plug. In addition, it activates Factors VIII, V, and Factor XIII, which forms covalent bonds that crosslink the fibrin polymers that form from activated monomers.
CONCLUSION
All the signs and symptoms that characterize transfusion related acute lung injury are explainable by neutrophils activation that through the production of extracellular traps with its component MPO and Elastase, cause direct toxicity on endothelial and ephitelial cells of the lungs, and through platelets activation induced by histones that causes microcirculatory injury due to the onset of clotting cascade which leads to a thrombus formation.

Formation of platelets-neutrophils aggregates promoted by NET, induces a positive feedback on neutrophils activation that increases extracellular traps creation.