DEFINITION
Pantothenate kinase-associated neurodegeneration, also known as neurodegeneration with brain iron accumulation (NBIA) and formerly called Hallervorden–Spatz Syndrome, is an autosomal recessive neurodegenerative disorder associated with iron accumulation in the nervous system. Its main clinical features include extrapyramidal dysfunction, early onset in childhood and a relentlessly progressive course.
Although the pathophysiologic basis of excessive iron accumulation and toxicity in PKAN is hypothetical at present, it has recently been shown that the disease may be caused by a defect in a novel pantothenate kinase gene.


Pantothenate kinase-associated neurodegeneration
EPIDEMIOLOGY
PKAN was first described by Drs. Julius Hallervorden and Hugo Spatz in 1922. Their discovery was brought about by a diagnosis of a family of 12 people in which five sisters exhibited progressively increasing dementia and dysarthria. Autopsies revealed brown discolorations in different areas of the brain (particularly of interest were, of course, the globus pallidus and substantia nigra regions).
However, reliable diagnosis of PKAN hit a milestone quite more recently with the availability of MRIs and genetic investigation.
Survival rates for those diagnosed with typical PKAN is 11.18 years with a standard deviation of 7.8 years. Prevalence data regarding this disorder remains incomplete, however it is estimated that anywhere between 1 in 1,000,000 to 3 in 1,000,000 individuals will be afflicted with this disorder (based upon observed cases in a population), but once again this is only an estimate as the disease is so rare it is difficult to statistically and accurately ascertain.
SYMPTOMS
Symptoms of PKAN typically begin in childhood and are progressive, often resulting in death by early adulthood.
The major symptoms include:
- Dystonia (repetitive uncontrollable muscle contractions that may cause jerking or twisting of certain muscle groups)
- Dysphagia and dysarthria due to muscle groups involved in speech being involved
- Respiratory compromise
- Rigidity/stiffness of limbs
- Tremor
- Writhing movements and seizures
- Dementia
- Spasticity
- Weakness
- Toe walking
- Retinitis pigmentosa, often resulting in a complete loss of vision.
25% of individuals experience an uncharacteristic form of PKAN that develops post-10 years of age and follows a slower, more gradual pace of deterioration than those pre-10 years of age. These individuals face significant speech deficits as well as psychiatric and behavioral disturbances.
Being a progressive, degenerative nerve illness, PKAN leads to early immobility and often death by early adulthood.
DIAGNOSIS
A neurological examination of the patient would show evidence of muscle rigidity, weakness, abnormal postures and tremors. If other family members are also affected, this may help determine the diagnosis.
Individuals exhibiting any of the above listed symptoms are often tested using MRI (Magnetic Resonance Imaging). On T2-weighted magnetic resonance (MR) images, PKAN patients reveal an eye-of-the-tiger sign: a marked low signal intensity circumscribes the globus pallidus, surrounding a central region of high signal intensity in the anteromedial globus pallidus. This effect is typically due to excess iron accumulation.


Generalized atrophy of the brain may also be noted, with a reduction in size of the caudate nuclei and tegmentum.
Microscopically, the characteristic changes include:
- Variable loss of neurons, myelinated fibers, and gliosis in globus pallidus and SN, which may appear spongiotic when severe
- Accumulation of pigment, ceroid lipofuscin and neuromelanin, mostly containing iron, in the globus pallidus
- Widely disseminated rounded or oval, nonnucleated structures called spheroids (also known as axon schollen or neuroaxonal dystrophy); these represent swollen axons with vacuolated cytoplasm and are found most abundantly in the pallidonigral system, although they are also present in the cerebral cortex
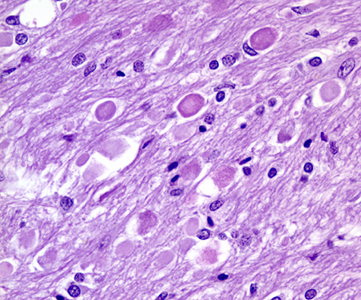
Iron deposition may be found intracellularly and extracellularly and frequently is centered on vessels. These changes are found to a lesser degree in other parts of the brain and in the spinal cord.
The presence of axonal spheroids suggests a link between PKAN and infantile neuroaxonal dystrophy. However, no clinical or genetic relationship has been reported between the two diseases. Also, other neuroaxonal dystrophies do not manifest excess iron deposition in the globus pallidus and would be expected to lack the eye-of-the-tiger sign at MR imaging. This could be a useful differentiating point since infantile neuroaxonal dystrophy may have a clinical manifestation similar to that of PKAN.
Development of diagnostic criteria continues in the hope of further separating PKAN from other forms of neurodegenerative diseases featuring NBIA.
POSSIBLE DIFFERENTIAL DIAGNOSIS OF PKAN
Wilson disease
Wilson disease usually presents with tremors, rigidity, dementia, and pseudobulbar features and has an autosomal recessive mode of inheritance. Examination of the eyes may reveal a Kayser-Fleischer ring, while MRI exhibits the characteristic changes consisting of high-intensity lesions in the basal ganglia, thalami, and mid brain. The normal low intensity of red nuclei and SN surrounded by abnormal high-signal intensity in the tegmentum of the mid brain gives rise to the typical face-of-the-giant-panda" sign. Results from serum ceruloplasmin and copper studies are usually abnormal and help to confirm the diagnosis. Neurologic symptoms are reversible if treated early with copper chelation therapy; hence, an early diagnosis is important.

Juvenile form of Huntington disease
The juvenile form of Huntington disease may be confused with PKAN, as patients with this form of Huntington disease present with a predominantly akinetic-rigid syndrome. The differentiating features include an autosomal dominant mode of inheritance and the presence of caudate atrophy on MRI.
Juvenile neuronal ceroid lipofuscinosis
Juvenile neuronal ceroid lipofuscinosis may also be difficult to distinguish from PKAN. It is an inherited disorder characterized by storage of ceroid and lipofuscin in neuronal and other tissues. The symptoms start in early childhood with vision loss, retinitis pigmentosa, dementia, rigidity, and dystonia. In contrast to the infantile and late-infantile forms of the disease, generalized tonic-clonic seizures and myoclonic seizures are not very common.
GM gangliosidoses
Rare metabolic disorders such as GM1 and GM2 gangliosidoses in children sometimes can have features similar to PKAN, but they have other clinical features and lab abnormalities and are differentiated readily.
PATHOGENESIS
PKAN is the first inborn error of pantothenate metabolism and the defects could result both from product deficit and a secondary excess of metabolites downstream of the block.
PANK2 mutations are predicted to result in CoA depletion and defective membrane biosynthesis in those tissues in which this is the major pantothenate kinase or in those tissues with greatest CoA demand.
In addition to this, as Acetyl-CoA is the main substrate for the Citric Acid Cycle , a CoA depletion will determine lowered Citrate levels. Being Citrate the essential substrate for active Aconitase, its depletion is predicted to cause Aconitase conversion in its cytoplasmic isoform IRP1. The activation of IRP1 may in turn lead to an increased ferritin synthesis, which could actively contribute to iron deposition in the brain.
In consideration of this, it may be possible to therapeutically deliver phosphopantothenate or an alternative intermediary compound to cells in the affected tissues in order to bypass the enzymatic defect and drive CoA synthesis.
As formerly stated, a mechanism of secondary metabolite accumulation could also be seen as the cause of high iron concentration in the brain. In fact, PANK is responsible for stifling the accumulation of N-pantothenoyl-cysteine and pantetheine. It is believed that when this accumulation is not suppressed, the result is direct cell toxicity or cell toxicity as a result of free radical damage due to the lack of suppression.
In normal brain, nonheme iron accumulates regionally and is highest in the medial globus pallidus and the substantia nigra pars reticulata, the two regions most severely affected in PKAN. Phosphopantothenate, the product of pantothenate kinase, normally condenses with cysteine in the next step in CoA synthesis (a cysteine is added to 4' - phosphopantothenate by the enzyme phosphopantothenoylcysteine synthetase to form 4' - phospho-N-pantothenoylcysteine). In PKAN phosphopantothenate is deficient, leading to Cysteine, N-pantothenoyl-cysteine and pantetheine accumulation.
Cysteine undergoes rapid auto-oxidation in the presence of iron, which results in free radical production and important cytotoxicity. In turn, iron-induced lipid peroxidation, a likely mechanism of secondary pathogenesis in PKAN, is enhanced by free cysteine, further stressing a cell with impaired membrane biosynthesis. Perturbed cysteine metabolism has been implicated in Parkinson and Alzheimer diseases as well. Thus, it has been proposed that pantothenate metabolism may represent a common pathway implicated in neurodegeneration.
Other references
PATIENT RISK FACTORS
Genetic
PKAN is an autosomal recessive disorder caused by a mutant PANK2 gene located at the chromosomal locus: 20p13-p12.3.
Mutant PANK2 gene-coded proteins are often caused by null or missense mutations, most notably a 7bp deletion in the PANK2 gene coding sequence.
The gene
PANK2 gene is normally responsible in coding for the protein Pantothenate kinase (PANK), the first enzyme in the biosynthetic pathway for Coenzyme A (which is particularly notable for its role in the synthesis and oxidation of fatty acids, and the oxidation of pyruvate in the citric acid cycle). More specifically, PANK phosphorylates pantothenate (vitamin B5) to form 4' - phosphopantothenate.

Some authors concluded that the symptoms observed in pantothenate kinase-associated neurodegeneration may be caused by a deficiency of the mitochondrial isoform of pantothenate kinase; they further postulated the existence of a complete intramitochondrial pathway for de novo synthesis of coenzyme A.
THERAPY
There have been no major breakthroughs in the treatment of PKAN, with most pharmacologic treatments focusing on the easing or temporary relieving of PKAN’s symptoms. Iron chelating agents have been used somewhat successfully in retarding the disorder, but they have not been a significant success.
Many believe taking certain vitamins may be beneficial, including pantothenate, Coenzyme Q, and other anti-oxidants.
Current research focuses on the future use of high dose pantothenate, the PANK2 enzyme substrate, in possibly alleviating symptoms as well as the further development of iron chelating agents that may be better aimed at reaching the central nervous system and working to better remove excess iron from the individual’s system.
Conclusion
Iron is normally absent from the central nervous system at birth, but iron deposits may occur physiologically throughout life in healthy adults, with the highest concentrations occurring in the globus pallidus (primarily in the form of the metalloprotein ferritin). This iron deposition is pathologically intensified in PKAN, while iron levels in blood and cerebrospinal fluid remain usually normal. Several histologic and radiologic studies have revealed that these iron deposits involve especially the basal ganglia and the globus pallidus.
In this respect, PKAN may serve as a model for complex neurodegenerative diseases, such as Parkinson disease, Alzheimer disease, Huntington disease and human immunodeficiency virus (HIV) encephalopathy, in which pathologic accumulation of iron in the brain is also observed. Thus, understanding the biochemical defect in this disease may provide key insights into the regulation of iron metabolism and its perturbation in this and other neurodegenerative diseases.