Sometimes it occurs that a very rare disease, such as the Fibrodysplasia Ossificans Progressiva, brings doctors to a deeper knowledge of some experimental treatments and helps to improve an existing method of diagnosis such as the mutational analysis. The Fibrodysplasia Ossificans Progressiva (FOP), also called "Munchmeyer disease" or "stone man syndrome" is a very rare genetic disorder characterized by an abnormal process of ossification. There is the presence of ectopic ossification in healing muscles, tendons and ligaments. Ossification can be spontaneous or wound-depending. Also, the surgical removal of the "new generated" bones has been shown to cause the body to "repair" the affected area with more bone.
CAUSES
The mutation is well known: FOP is caused by an autosomal dominant allele with complete penetrance on chromosome 2p23-24. As the fitness is highly reduced, the disease is mostly caused by spontaneous mutation in the gamete. It affects about 1 in every 2 million people (approximately 30 patients only in Italy). The gene responsible for the disease is ACVR1, which encodes the activin receptor, a BMP type-1 receptor. Codon 206 is changed from arginine to histidine. In people carrying this mutation, endotelial cells are likely to transform into mesenchymal stem cells and then into bone.
Wikipedia, the free encyclopedia
SYMPTOMS


Because it is very rare, the disease and its symptoms are often misdiagnosed as cancer or fibrosis. The newborns with FOP have deformed big toes or in some cases a notable lump on their body. The first "flare-up" that leads to the formation of new and abnormal bones occurs before the age of 10. Bone formation follows the same order of the fetus: from the top of the body downward. However, it does not necessarily occcur in this order due to injury-caused flare-ups. All the new bones grow independently from the normal skeleton but they can easily fuse with the existing bones. All the muscles, at last, turn into bones, except the tongue, extra-ocular muscles and cardiac and smooth muscles. Joints start to remain locked, and death occurs when the chest can't move on any more to breath-in and out, due to the bone tissue that covers it.
The gene that causes ossification is normally deactivated after a fetus' bones are formed in the womb, but in patient with FOP it keeps working after birth. Patients should pay attention not to damage their body with any injury (by for example to a fall or a cut) because it is the inflammatory response that triggers the formation of new bone. Injuried cells incorrectly express an enzyme for bone repair during their apoptosis, resulting in lymphocytes containing too much BMP4 (Bone Morphogenetic Protein 4). This protein is a product that contributes to the development of the skeleton in the normal fetus. Another feature of FOP is the hearing impariment, which occurs in approximately 50% of patients. The onset is usually in childhood or adolescence and is generally slowly progressive. Hearing loss is usually conductive in nature and may be result of middle-hear or acoustic meatus ossification, but strangely in some patients the hearing impairment is neurological in nature. Other more variable features of FOP are a diffuse scalp baldness and, for girls, menstrual irregolarities and premature menopause. In some patients, even if not in all of them, FOP can be exacerbated during growth spurts and puberty. Longitudinal growth seems to be normal although severe scoliosis may develop expecially in cases with an asymmetrical heterotopic ossification of the spine. Sexual development is usually normal, although lack of breast development in females has been noted. Pregnancy in patients with FOP is full of risks and there is a probability of 0,5 to give birth to a FOP baby if only the mother or the father is affected: first of all, in fact, limitations in posture and joint mobility may place the woman's or the fetus' health at risk in the case of pregnancy; second, FOP is an autosomal dominant disorder with full penetrance but variable expression. Genetic counseling and guidance regarding issues of contraception are warranted for the sexually active or those who are considering such activity.
THE FOP LESION

As written before, clinically the lesions in FOP (even if not all of them) are wound-depending or, in other words, they are induced by injuries. They are characterized by painful swellings in soft tissues that progress to form mature heterotopic bone. The histological stages of lesion formation are well described and include, in order of progression:
* Perivascular lymphocytic infiltration
* Lymphocytic migration into affected muscle and myonecrosis
* Early reactive fibroproliferation
* Intense fibroproliferation
* Neovascularity and angiogenesis
* Cartilage formation
* Endochondral bone formation
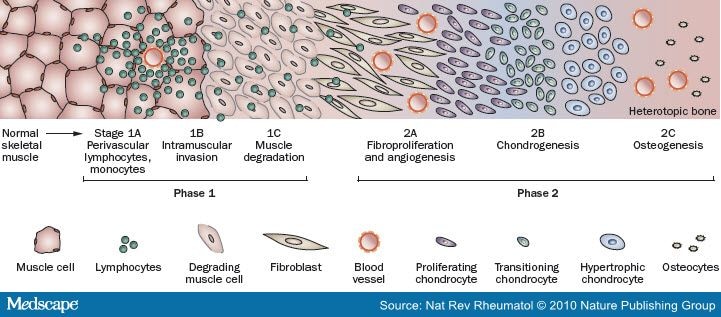
Early FOP lesions contain an intense perivascular B- and T-cell lymphocytic infiltrate. An intense fibroproliferative reaction followed by neovascularity and angiogenesis is characteristically found in the next stages. These stages, unfortunately, are microscopically indistinguishable from aggressive juvenile fibromatosis, and this can easily lead to a misdiagnosis. Then, the new fibroproliferative tissue transform into cartilage and then into bone. The entire process, as shown above, has been studied from a hostological point of view. All stages of histological development are present in the FOP lesion within days of its induction, indicating that different regions mature at different rates. Although heterotopic bone formation in FOP is similar in some respects to bone formation in embryonic skeletal development and postnatal fracture healing, important differences are the lack of inflammation in embryonic skeletal induction and the relative absence of lymphocytic inflammatory cells in early fracture healing. Dorsal, axial, cranial and proximal areas of the body tend to be involved first, with ventral, appendicular, caudal and distal areas involved later. Also, radiographic evidence supports the notion that the heterotopic skeleton in FOP has macroscopic features of modeling and remodeling similar to those found in the normotopic skeleton. New bones have a mature cortical and trabecular organization and even the presence of well-defined corical-endosteal borders enclosing medulary canals.
THORACIC INSUFFICIENCY SYNDROME

One of the complications of the FOP, common in most patients, is, of course, the thoracic insufficiency syndrome (TIS) which can lead to life-threatening complications. TIS is the inability of the chest to support normal respiration or lung growth, and it is seen in patients who has severe restrictive disease of the chest wall that is fused with ribs. The orthotopic ankylosis of the costovertebral joints, the ossification of the intercostal muscles and other spinal and costovertebral deformities can contribute to TIS. These kinds of malformations are common in patients with FOP and mostly develop early in life, at the age of 2-7 years old. The restrictive disease is also shown in children before the first flare-ups of the heterotopic ossification. TIS is also related with pneumonia and right-sided congestive heart failure. Often, the primary changes in FOP are in the connective tissues like aponeuroses, fasciae and tendons scattered on the spine. During the first decade of life, soft-tissue swelling similar to small tumors seize the aponeuroses, fasciae, tendons and muscles of the back, slowly transforming all these tissues into bone through and endochondral process. Eventually, they make costovertebral joints locked in place, rendering movement impossible. Many patients with TIS have, according to the restrictive disease, abnormal spirometry and also abnormal electrocardiograms expecially in old patients. The electrocardiographic evidence of right ventricular disfunction is then highly related with TIS and FOP: patients can easily die of right-sided heart failure. People with FOP also tend to retain carbon dioxide and are at high risk of sudden respiratory failure and death during unmonitored use of oxygen. In addiction, bacterial infections such as pneumonia are very common and they represent one of the most importan causes of death in FOP (as in the case of Harry Eastlack). That is why the median life span of patients with FOP is approximately 45 years.
IMMUNOLOGICAL FEATURES
The dysregulation of the BMP4 signaling pathway can explain many other features of FOP. In fact, there are also underlying immunological components of the disorder due to the role of BMP4 in the regulation of the immune system. Many flare-ups triggered by virus and immunizations and influenza-like prodrome have been shown many times in patients with FOP. During heterotopic ossification, lymphocytes seem to infiltrate and destruct skeletal muscles and the whole process of ossification can be partially paused by the use of immunosoppressive agents or corticosteroids. In other words, the immune system plays a prominent and provocative role in the pathophysiology of the disease. BMP4 plays a presumptive role in both B-cells and T-cells development during embryogenesis. BMP signals are both required for the formation of the thymus (they specifically control the thymic microenvironment) and of the microenvironment in the bone marrow. Talking about BMP4, it seems to regulate thymic homeostasis and T-cell lineage differentiation, as far as we know from recent experiments. Also, the finding that BMPs are expressed in peripheral immune cells indicates that BMPs function beyond T-cell development, and suggests that they are likely key regulators of the adaptive immune system. As most mices deficient in elements of the BMP-signaling pathway have severe and complicated phenotypes leading to premature death, functional data to support this notion are largely missing and our understanding is therefore very incomplete.
ENVIRONMENTAL FACTORS IN FOP: THE STUDY OF MONOZYGOTIC TWINS
As about in every genetic disorder, the phenotype of FOP is affected both by genetic mutations and environmental factors. Studying the role of environment is not easy when talking about a very rare disease and the only solution is to study monozygotic twins. There are only three known pairs of MZ twins with FOP, and all three were studied by scientists who examined their anamnesis and also a follow-up of three years. They found that, within each pair, congenital toe malformations were identical. However, postnatal heterotopic ossification varied greatly depending on life history and, of course, environmental exposure. It is now clear that genetic determinants strongly influence disease phenotype during prenatal development and that environmental factors strongly influence postnatal progression of the disease. In fact, results clearly showed that the location and intensity of each flare-up is different even within a pair of MZ twins, depending on surgical operations and injuries differently received during life. Recent studies on twins genetic background also underlined that the difference of intensity in flare-ups can be related to the differences in the twins' immune system. Although MZ twins are considered to have identical genomes, there is one organ system, the immune system, where the genomes are invariably different. This is especially true at loci that encode the B-cell and T-cell receptors, as well as the immunoglobulin loci where genetic rearrangements are of paramount importance in establishing immunological diversity. This feature of genetic diversity, even among MZ twins, may have particular relevance for patients with FOP because immunological factors are believed to play a role in the progression of the disease.
Wikipedia, the free encyclopedia
Clinical reviews in bone and mineral metabolism
LABORATORY FINDINGS IN FOP
Routine biochemical evaluations of bone mineral metabolism are usually normal, although bone remodeling rates and alkaline phosphatase activity in the serum may be increased, especially during disease flare-ups. Urinary basic fibroblast growth factor levels may be elevated during disease flare-ups, and coincide with the preosseous angiogenic phase of fibroproliferative lesions. Nephrolithiasis is more common in older patients with FOP and may be caused by increased immobilization and dehydratation in the setting of generalized increased bone remodelling and miner turnover.
Clinical reviews in bone and mineral metabolism
CASES
From history, many people who apparently "turned to stone" are from legends said to have existed. The best known case is that of Harry Eastlack (1933-1973). His first symptoms appeared at the age of 10 and, when he died of pneumonia at the age of 40, his very entire body has completely turned to bone, leaving him only the opportunity to move his lips. After death, he donated his skeleton to science, in order to seek and find a proper treatment for his condition. His skeleton is shown in the photo above.
Clinical reviews in bone and mineral metabolism
EARLY DIAGNOSIS AND MUTATIONAL ANALYSIS
A correct and quick diagnosis is the most important thing when trying to treat a rare and destabilizing disease like FOP. An early diagnosis, in fact, reduce the risk of surgical iatrogenic damage and allows us to try to reduce and slow down the effects of this condition. Unfortunately, most patients with FOP are misdiagnosed early in life (mostly before the onset of the heterotopic ossification) and undergo dangerous diagnostic and surgical procedures that easily cause lifelong disability, due to the surgical wounds that trigger the process of ossification. Actually, there are two ways to diagnose FOP: the first one is the physical examination; the second one is the mutational analysis Each patient needs both for a correct diagnosis.
The physical examination is concentrated in the analysis of the skeleton and soft tissues: most of the children show , as written before, congenital malformations of the great toe. There's also the chance to find some soft tissue lesions of the neck and the back (often as a result of invasive procedures due to a misdiagnosis) that clearly are early flare-ups of the disease. But the "gold standard" for Fibrodyspasia Ossificans Progressiva is, of course, the DNA sequence analysis, that looks for the missense mutation, a single nucleotide substitution in the 206 codon of the ACVR1 gene (mutation C617->A).
This analysis is called mutational analysis, and was recently improved by a team of Italian physicians, who found another mutation related with FOP.
h3. GENETIC DISORDERS
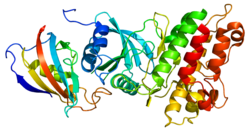

Speaking from the genetic side, as the reproductive fitness is very low in patients with FOP, this condition is sporadic and it is due to a spontanous mutation. However, some familial cases have been described with an autosomal dominant pattern of inheritance and variable expression. The candidate gene is ACVR1. This gene, as said before, encodes the activine A type 1 receptor, a receptor for BMPs. It is basically a serine/threonine receptor kinase belonging to the TGFB-R family. The receptors contain an extra-cellular ligand-binding domain, a single transmembrane domain, a cytoplasmic serine/threonine kinase domain and also an additional regulatory domain that is not present in type II receptors. This region, called GS domain, has an important role in FOP pathogenesis. The GS domain is phosphorylated by type II receptors to trigger the signaling cascade inside the cell after receptor-binding by ligand. The phosphorylation cascade activates the SMAD proteins that modulate the expression of specific target genes. The GS domain, well conserved, has an important regulatory role for the entire receptor. It binds FKBP12 (the FK506 binding protein 12). FK506 is a protein that can induce osteogenesis in combination with BMPs. When FKBP12 binds the GS domain, it keeps the receptor inactivated, preventing leaky signaling in the absence of ligand (that is, no osteogenesis). However, FK506 can remove FKBP12 from the receptor, which is then able to promote osteogenesis. Using a 3d model of the homologous TGFBR-1, it was possible to show that the basic arginine residue, R206, included in the GS domain, forms a salt bridge with an invariant aspartate residue that links the GS domain to a SMAD specificity site. The substitution of the arginine 206 with histidine in FOP is then responsible of a conformational change in the GS domain, that is no longer able to bind FKBP12: this leads to a ligand-independent activation of the receptor.
The Italian team carried out a mutational analysis on 17 Italian FOP patients. The DNA was extracted from peripheral blood samples of the patients and, in some cases, of the parents. The process of sequencing was carried out on the DNA and also on the RNA extracted from lymphocytes, after a retrotranscription with rt-PCR. They used two specific primers to isolare the ACVR1 gene. 15 patients were heterozygous for the classic mutation in ACVR1, but the other two resulted negative. One of them was carrying a new variant mutation in the R258 residue of ACVR1 (c774G>C). This patient had the absence of great toe malformation, but an early onset of the flare-ups at the age of 4. This newly identified mutation is a substitution of another highly-conserved residue. This residue is also a surface residue, as the one of the "classic" mutation. Unlike R206, R258 is not part of the GS domain; however, it is packed against the GS loop, and can be involved in determining the position of the GS domain. So, in both mutations it is a conformational change in the GS domain that abnormally activates the signaling: in this case if the GS domain is less firmly anchored to the remaining part of the protein, it is more likely to be phosphorylated and activated. Both substitutions, in fact, are gain-of-function mutations. Strangely, even if the genetic alterations are only limited, as far as we know, to these two mutations, there is a great variability in the severity of the disease, including differences in the age on onset, frequency, location and duration of flares-up and so on. This is the very evidence that environmental factors also can affect the phenotypic variability.
Mutational analysis of the ACVR1 gene in Italian patients affected with fibrodysplasia ossificans progressiva: confirmations and advancements
PROPHYLACTIC ISSUES IN FOP
Of course even in a disease like FOP, prevention is better than cure. All dangerous behaviours (such as extreme sports, harmful activities etc...) should be avoided by the patients in order to prevent every form of wound-depending flare-up and heterotopic ossification. Tattoes, piercings and other kinds of voluntary injuries should be avoided as well. Dental therapy must involve assiduous attention to prophylaxis of caries and must preclude intramuscular injection of local anesthetics, especially mandibular blocks and the stretching of the jaw. All intramuscular injections must be avoided: a general anesthesia should be preferred. Prevention of falls is crucial. Prophylaxis against influenza and pneumonia, as well as measures to prevent respiratory infection and cardiopulmonary complications of restrictive chest-wall disease, are vitally important. Every kind of biopsy and every invasive surgical intervention shoul be avoided as well, when possible.
Clinical reviews in bone and mineral metabolism
TREATMENT
There is no existing cure for FOP. Every attempt to surgically remove the bone results in more robust bone growth. Considering the paucity of patients afflicted with the disorder, the erratic natural history of the disease and the extreme variability of FOP, double-blinded, randomized and controlled studies are extremely difficult to conduct in the FOP community. The current treatment recommendations have largely been made based on anecdotal data or poorly controlled clinical studies. Because of the difficulty in designing clinical trials, a clinically relevant animal model would provide a potential testing platform for studying drug development. Unfortunately, there are no naturally occurring animal models of heterotopic ossification that accurately reproduce all of the clinical features of FOP. Heterotopic ossification can be induced in an animal by the injection, surgical implantation, or genetic overproduction of BMPs. These animal models, however imperfect, can help accelerate the pace of drug development and preclinical testing. Recently, following these guidelines, scientists are discovering new possible treatments, that are still in the experimental stage. There are three main possible treatments, unfortunately still without clinical trials:
# The use of Immunomodulators, following the hypotesis of the "immune dysregulation" disease. The suppresive effects on lymphocytes of many corticosteroids may help to reduce the intense lymphocytic infiltration and tissue edema seen in the early stages of the disease. Steroids are most effective when used early to treat discrete flare-ups. In addition, Mast-Cell Inhibitors, Nonsteroidal Anti-Inflammatory Drugs and COX-2 Inhibitors can properly reduce the inflammatory response in order to soften the negative effects and the number of flare-ups, expecially when these drugs are used early in life;
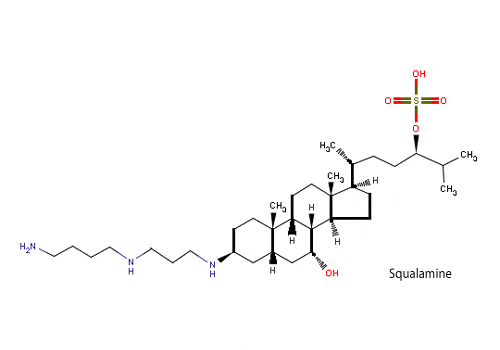
# The use of anti-angiogenic agents: angiogenesis is an absolute requirement for the fomation and development of heterotopic bone. BMPs are potent inducers of ID-1 expression, a gene required for angiogenesis. The goal of anti-angiogenic therapy in FOP is to inhibit new blood vessel formation in order to slow down or inhibit the subsequent production of new bone formation once a new lesion has appeared. So, angiogenesis may potentially be minimized with anti-angiogenic agents, such as nonsteroidal anti-inflammatory drugs, COX-2 inhibitors, thalidomide and vascular growth factor traps. In addition, the use of Squalamine, an antiangiogenic aminosterol, can prevent the growth of blood vessels in cartilagineous tissue, in this way preventing the creation of new bone. This aminosterol is active in sharks that in fact have a cartilagineous skeleton, bone-free;
 # BMP antagonists, BMP receptor antibodies and signal transduction inhibitors: irrespective of the regulation of the BMP signaling mediated through binding to its receptors remains a critical step in the induction of abnormal ossification. BMP4-induced heterotopic ossification can be prevented by local delivery of wild-type Noggin or after somatic cell gene transfer of the Noggin or after somatic cell gene transfer of the Noggin variant. Noggin gene therapy continues to be promising long-term treatment for FOP based on our present knowledge of the condition. Before gene therapy can become a clinical reality, methods must be developed for safety delivering Nogging to target cells and for regulating Noggin gene expression at those cellular targets. Additional treatment options to consider for the future include the use of monoclonal antibodies directed against the overabundant cell surface BMP receptors, and signaling transduction inhibitors directed against the dysregulated BMP signaling pathway in FOP;
# BMP antagonists, BMP receptor antibodies and signal transduction inhibitors: irrespective of the regulation of the BMP signaling mediated through binding to its receptors remains a critical step in the induction of abnormal ossification. BMP4-induced heterotopic ossification can be prevented by local delivery of wild-type Noggin or after somatic cell gene transfer of the Noggin or after somatic cell gene transfer of the Noggin variant. Noggin gene therapy continues to be promising long-term treatment for FOP based on our present knowledge of the condition. Before gene therapy can become a clinical reality, methods must be developed for safety delivering Nogging to target cells and for regulating Noggin gene expression at those cellular targets. Additional treatment options to consider for the future include the use of monoclonal antibodies directed against the overabundant cell surface BMP receptors, and signaling transduction inhibitors directed against the dysregulated BMP signaling pathway in FOP;
# The use of the RNA interference, that can silence the damaged copy of the gene using an engineered RNA molecule, can restore the cellular function damaged by FOP mutation. Cells, in this way, keep the bad copy of the gene, but all the mRNA of that gene is destroyed (the gene is silenced). Only the normal copy of ACVR1 keeps working. This is similar to gene therapy, but without the use of any virus or DNA, it seems to be much safer. Since RNAi may not totally abolish expression of the gene, this technique is sometimes referred as a "knockdown", to distinguish it from knockout procedures in which expression of a gene is entirely eliminated.
Current approaches to FOP treatment are palliative and symptom-modifying. A continued rigorous approach to understanding the underlying genetic cause and molecular pathophysiology of the condition offer the best hope for designing effective treatments and preventions thart are truly disease-modifying.
Clinical reviews in bone and mineral metabolism
Rna and fop
AN INNOVATION: RNA INTERFERENCE

The use of RNAi is the most innovative possible treatment. Scientists like Eileen M. Shore and Frederick Kaplan, at the University of Pennsylvania, supported by the National Institute of Arthritis and Musculoskeletal and Skin Diseases, have described in a recent issue of Gene Therapy how a process like RNAi restored normal cell-signaling in the cells from patients with FOP. The disease is an ideal target for the RNAi treatment because it is an autosomal dominant disorder, which means that only one allele of the gene is defective. If this allele is silenced, as written before, the other one can work properly even if alone. Following this hypotesis, Josef Kaplan created a short RNA molecule engineered in order to bind to the mutated region of the ACVR1 mRNA. This would silence only the defective allele. To test the effects of this RNA, the team carried out some clever experiments on cultured cells taken from the baby teeth of children with FOP. This cells are ideal for this kind of experiments because they can normally turn to bone and they do not require a biopsy to extract since baby teeth fall out naturally. The results were all succesfull: the RNAi actually silenced the mutated gene restoring the right signal pathways. The University of Pennsylvania team's next step is then to develop a mouse model of FOP in order to repeat these experiments. If they are as succesfull as the previous ones, a clinical trial of the treatment will be required.
CONCLUSION
As in every rare genetic disorder with no available cure, our efforts regarding FOP have to be focused on an early diagnosis and a proper treatment. Even if it is so rare, a clinical trial carried out by many Countries should become a reality. If not, it will be always hard to find a cure only studying the medical cases of the past or tiny cells in a laboratory. We already moved a step forward using animal models of the disease testing the first possible treatments, and this is a piece of good news, meaning that the next step will be the first clinical trial. RNAi seems to be the proper treatment not only in order to cure FOP, but also to cure other autosomal dominant genetic disorders such as Huntington's disease or even cancer. Patients should be the first subjects of our studies and their benefits should be, as ever in medicine, our first goal.