Definition
The Blue Skin Syndrome is a rare congenital desease, also called methemoglobinemia.
Methemoglobinemia (or methaemoglobinaemia) is a disorder characterized by the presence of a higher than normal level of methemoglobin in the blood.
Methemoglobin is a form of hemoglobin that contains ferric [Fe3+] iron and has a decreased ability to bind oxygen. However, the ferric iron has an increased affinity for bound oxygen. The binding of oxygen to methemoglobin results in an increased affinity of oxygen to the three other heme sites (that are still ferrous) within the same tetrameric hemoglobin unit. This leads to an overall reduced ability of the red blood cell to release oxygen to tissues, with the associated oxygen–hemoglobin dissociation curve therefore shifted to the left. When methemoglobin concentration is elevated in red blood cells, tissue hypoxia can occur.


(Introduction to methemoglobinemia)
Pathophysiology
Red blood cells contain hemoglobin, which has a quaternary structure. Each hemoglobin molecule is composed of 4 polypeptide chains. Each of these chains is associated with a heme group, which contains iron in the reduced or ferrous form (Fe2+). In this form, iron can combine with oxygen by sharing an electron, thus forming oxyhemoglobin. When oxyhemoglobin releases oxygen to the tissues, the iron molecule is restored to its original ferrous state. Hemoglobin can accept and transport oxygen only when the iron atom is in its ferrous form. When hemoglobin loses an electron and becomes oxidized, the iron atom is converted to the ferric state (Fe3+), resulting in the formation of methemoglobin. Methemoglobin lacks the electron that is needed to form a bond with oxygen and thus is incapable of oxygen transport.
Under normal conditions, methemoglobin levels remain below 1%; however, under conditions that cause oxidative stress, levels will rise.
The low level of methemoglobin is maintained through two important mechanisms:
- The first is the hexose-monophosphate shunt pathway within the erythrocyte. Through this pathway, oxidizing agents are reduced by glutathione.
- The second and more important mechanism involves two enzyme systems, diaphorase I and diaphorase II, and requires nicotinamide adenine dinucleotide (NADH) and nicotinamide adenine dinucleotide phosphate (NADPH), respectively, to reduce methemoglobin to its original ferrous state.
NADH-dependent methemoglobin reduction (diaphorase I pathway) is the major enzymatic system involved. Cytochrome b5 reductase plays a major role in this process by transferring electrons from NADH to methemoglobin, an action that results in the reduction of methemoglobin to hemoglobin. This enzyme system is responsible for the removal of 95-99% of the methemoglobin that is produced under normal circumstances.
NADPH-dependent methemoglobin reduction (diaphorase II pathway) usually plays only a minor role in the removal of methemoglobin. This enzyme system utilizes glutathione production and glucose-6-phosphate dehydrogenase (G6PD) to reduce methemoglobin to hemoglobin. It assumes a larger and more important role in methemoglobin regulation in patients with cytochrome b5 reductase deficiencies.
Disruptions with these enzyme systems lead to the condition.

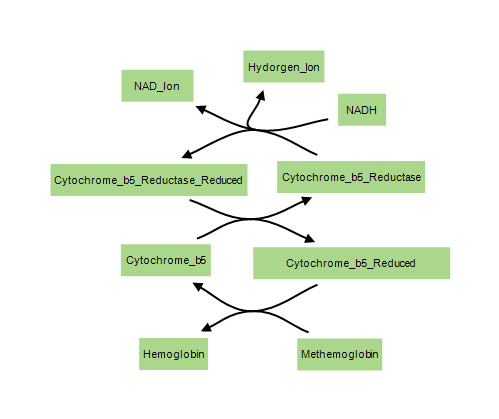
Elevated levels of methemoglobin in the blood are caused when these mechanisms are overwhelmed and the oxygen carrying ferrous ion (Fe2+) of the heme group of the hemoglobin molecule is oxidized to the ferric state (Fe3+). This converts hemoglobin to methemoglobin, resulting in a reduced ability to release oxygen to tissues and thereby hypoxia. This can give the blood a bluish or chocolate-brown color.
Hypoxia occurs due to the decreased oxygen-binding capacity of methemoglobin, as well as the increased oxygen-binding affinity of other subunits in the same hemoglobin molecule which prevents them from releasing oxygen at normal tissue oxygen levels.
(Pathophysiological mechanisms of Methemoglobinemia)
Types
There are two forms of Methemoglobinemia:
- inherited or congenital
- acquired (by exposure to certain drugs, chemicals, or foods)
Congenital methemoglobinemia
There are two forms of inherited methemoglobinemia.
- The first form is passed on by both parents. The parents usually do not have the condition themselves, but they carry the gene that causes the condition. It occurs when there is a problem with an enzyme called NADH-cytochrome b5 reductase 3 (CYB5R3, also known as Diaphorase-1 and NADH-methemoglobin reductase).
This enzyme is a flavin adenine dinucleotide (FAD) containing enzyme catalyzing electron transfer from NADH to cytochrome b5. A single gene located on chromosome 22q13
(GeneID: 1727) produces two protein variants:
- A soluble form is present predominantly in erythrocytes
- A membrane-bound form is localized to the endoplasmic reticulum (ER), mitochondria, nuclei, peroxisomes and plasma membranes of somatic cells.
Deficiency of CYB5R3 (a rare autosomal recessive disorder) leads to hereditary methemoglobinemia.
It is present as two distinct phenotypes, type I and type II:
- Type I methemoglobinemia (also called erythrocyte reductase deficiency) is characterized by CYB5R3 enzyme deficiency restricted to erythrocytes and is associated with benign cyanosis.
- Type II methemoglobinemia (also called generalized reductase deficiency) is associated with generalized CYB5R3 deficiency affecting all cells and is characterized by mild cyanosis with severe neurologic disability and numerous developmental defects the affected infants usually die at an early age. In this case the enzyme doesn't work anywhere in the body.
Identification and characterization of mutation in Type I methemoglobinemia:


(Molecular Basis of Two Novel Mutations Found in Type I Methemoglobinemia, 2011 )
- The second form of inherited methemoglobinemia is called hemoglobin M disease. It is caused by defects in the hemoglobin molecule itself. Infact, in most of the hemoglobin M, tyrosine has been substituted for either the proximal or the distal histidine. This results in reduced capacity of the enzymatic machinery of the erythrocyte to efficiently reduce the iron to the divalent form and thus predisposes to methemoglobinemia. There are three phenotypic varieties of Hb M, corresponding to the globulin gene affected (alpha, beta, or gamma) as follows:
• Alpha-chain variants cause neonatal cyanosis that is persistent
• Beta-chain variants do not cause cyanosis until several months after birth, when the level of fetal hemoglobin has decreased
• Gamma-chain variants cause transient neonatal cyanosis that resolves once the level of fetal hemoglobin decreases
This disorder is inherited in an autosomal dominant pattern: only one parent needs to pass on the abnormal gene for the child to inherit the disease.
(West J Med. 2001 )
Acquired methemoglobinemia
Methemoglobinemia can also be acquired (more common form). The protective enzyme systems normally present in red blood cells maintain methemoglobin levels at less than one percent of the total hemoglobin in healthy people. Exposure to exogenous oxidizing drugs and their metabolites (such as benzocaine, benzene, certain antibiotics and nitrates) may accelerate the rate of formation of methemoglobin up to one-thousandfold, overwhelming the protective enzyme systems and acutely increasing methemoglobin levels.

Symptoms
Methemoglobinemia may be acute or chronic. The physiologic level of methemoglobin in the blood is 0% to 2%.2 Methemoglobin concentrations of 10% to 20% are tolerated well, but levels above this are often associated with symptoms. Levels above 70% may cause death. Symptoms also depend on the rapidity of its formation. Many patients with lifelong methemoglobinemia are asymptomatic, but patients exposed to drugs and toxins who abruptly develop the same levels of methemoglobinemia may be severely symptomatic.
Signs and symptoms of methemoglobinemia (methemoglobin >1%) include:
- shortness of breath,
- cyanosis,
- mental status changes (~50%),
- headache,
- fatigue, exercise intolerance,
- dizziness,
- loss of consciousness.

Arterial blood with elevated methemoglobin levels has a characteristic chocolate-brown color as compared to normal bright red oxygen-containing arterial blood.
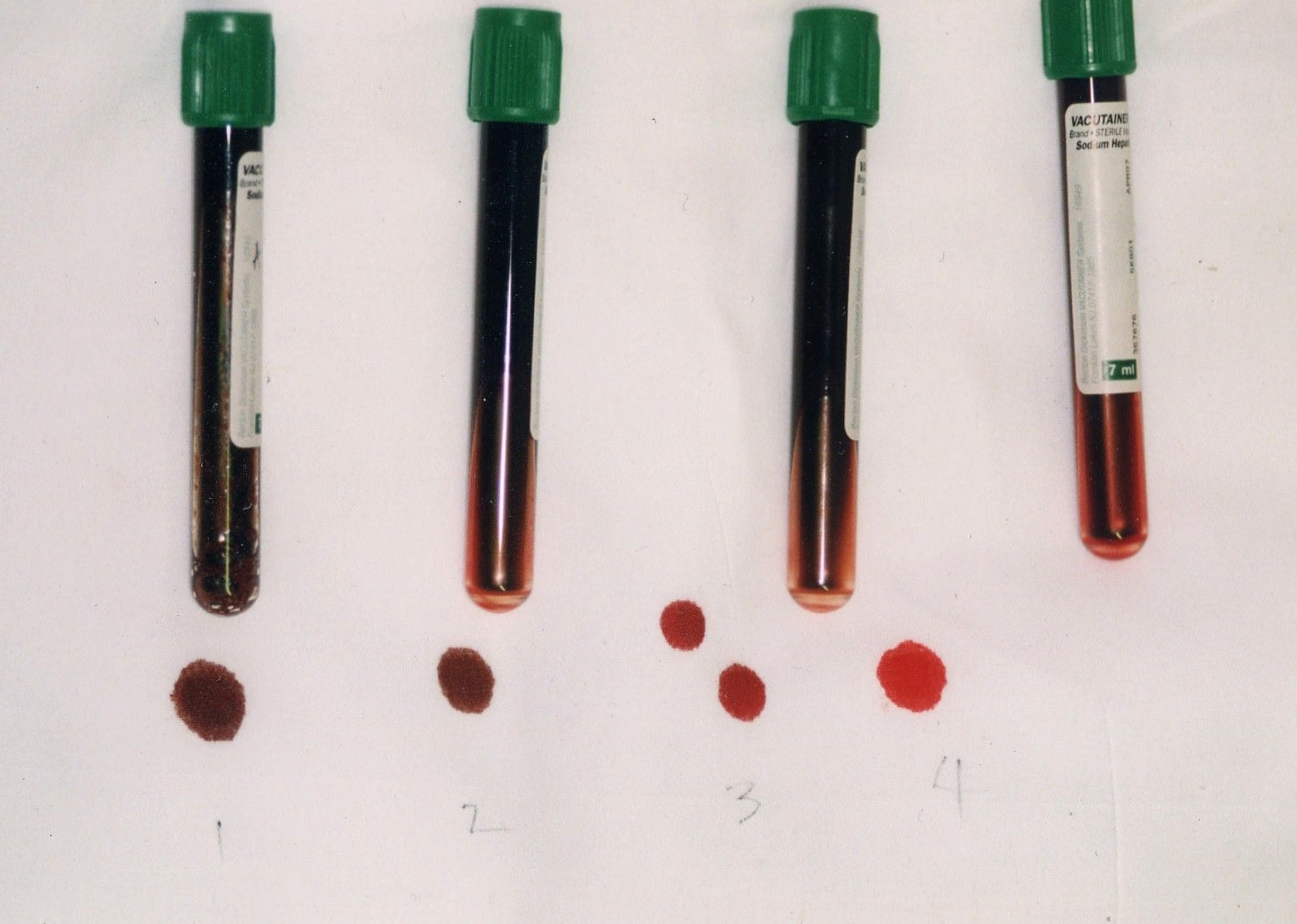
Cyanosis and characteristic color of blood determinate bluish coloring of the skin.
However, patients with co-morbidities such as anemia, cardiovascular disease, lung disease, sepsis, or presence of other abnormal hemoglobin species (e.g. carboxyhemoglobin, sulfehemoglobin or sickle hemoglobin) may experience moderate to severe symptoms at much lower levels (as low as 5-8%).
(Methemoglobinemia: from diagnosis to treatment, 2008)
(Methemoglobinemia Clinical Presentation,2013)
Diagnosis
Methemoglobinemia can be diagnosed with a blood test.
Blood containing high concentrations of methemoglobin appears chocolate brown. Subjects with methemoglobinemia may have normal partial pressures of oxygen, despite life-threatening methemoglobinemia. The oxygen saturation values, measured by a pulse oximeter, are falsely elevated.
In methemoglobinemia due to drug exposure, an elevated level of methemoglobin is found, but the activity of NADH cytochrome b5 reductase is normal. In hereditary type II methemoglobinemia, the enzyme's activity is less than 20% of normal. Hemoglobin M may be differentiated from methemoglobin formed from hemoglobin A by its absorption spectrum in the range of 450 to 750 nm. Electrophoresis at pH 7.1 is most useful for the separation of hemoglobin M.
(How do you confirm the diagnosis?)
Treatment
The course of hereditary methemoglobinemia type I is benign, but these patients should not be administered oxidant drugs. Treatment may be required for cosmetic reasons or for an inadvertent use of oxidant drugs. Ascorbic acid, 300 to 600 mg orally daily divided into 3 or 4 doses, is helpful.
For methemoglobinemia due to drug exposure, traditional first-line therapy consists of somministration od supplemental oxygen and an infusion of methylene blue (1 to 2 mg/kg intravenously slowly over five minutes followed by IV flush with normal saline), whose action depends on the availability of reduced nicotinamide adenine nucleotide phosphate (NADPH) within the red blood cells.
After an acute exposure to an oxidizing agent, treatment should be considered when the methemoglobin is 30% in an asymptomatic patient and 20% in a symptomatic patient.
Patients with anemia or cardiorespiratory problems should be treated at lower levels of methemoglobin.
Methemoglobinemia due to hemoglobin M does not respond to ascorbic acid or methylene blue.
Dextrose should be given because the major source of NADH in the red blood cells is the catabolism of sugar through glycolysis. Dextrose is also necessary to form NADPH through the hexose monophosphate shunt, which is necessary for methylene blue to be effective.
Methylene blue is an oxidant; its metabolic product leukomethylene blue is the reducing agent. Note: It may be dangerous in patients who have or may be at risk for a blood disease called G6PD deficiency, and should not be used. Infact patients with G6PD deficiency also may not produce sufficient NADPH to reduce methylene blue to leukomethylene blue.

Alternative treatments include hyperbaric oxygen therapy and exchange transfusions.
In most cases of mild acquired methemoglobinemia, no treatment is needed. However, you should avoid the medicine or chemical that caused the problem. Severe cases may need treatment, which may include a blood transfusion.
(What treatment works?)
Prognosis
People with type 1 methemoglobinemia and hemoglobin M disease usually do well. Patients are usually asymptomatic, except for the presence of chronic cyanosis.
Type 2 methemoglobinemia is much more serious, and usually causes death within the first few years of life, primarily because of multiple neurologic complications.
Acquired methemoglobinemia is usually mild but may be severe and rarely fatal, depending on the cause. This people usually do very well once the drug, food, or chemical that caused the problem is identified and avoided.
Possible Complications
- Shock
- Myocardial infarction
- Seizures
- Coma and Death
(Outlook and possible problems)
Prevention
Patients with inherited methemoglobinemia should be counseled regarding the avoidance of toxins, chemicals, and certain drugs (eg, dapsone).
Genetic counseling is recommended for couples with a family history of methemoglobinemia who are considering having children.
Carriers
The Fugates, a family that lived in the hills of Kentucky, are the most famous example of this hereditary genetic condition. They are known as the "Blue Fugates". Martin Fugate settled near Hazard, Kentucky, circa 1800. His wife was a carrier of the recessive methemoglobinemia (met-H) gene, as was a nearby clan with whom the Fugates intermarried. As a result, many descendants of the Fugates were born with met-H.
The "blue men of Lurgan" were a pair of Lurgan men suffering from what was described as "familial idiopathic methaemoglobinaemia" who were treated by Dr. James Deeny in 1942. Deeny, who would later become the Chief Medical Officer of the Republic of Ireland, prescribed a course of ascorbic acid and sodium bicarbonate. In case one, by the eighth day of treatment there was a marked change in appearance and by the twelfth day of treatment the patient's complexion was normal. In case two, the patient's complexion reached normality over a month-long duration of treatment.


(Blue men are real)