INTRODUCTION
Among the hereditary sensory and autonomic neuropathies (HSAN), type IV (HSAN IV), also known as congenital insensitivity to pain with anhidrosis (CIPA; MIM 256800), is a rare autosomal recessive disorder.
CIPA is characterized by the absence of reaction to noxius stimuli, the inability to sweat, hyperpyrexia, mild retardation, and sel-mutilating behavior. Thus the name congenital, meaning present of birth; insensitivity to pain,meaning inability to feel pain; anhidrosis, the inability to sweat. This inability causes the hyperpyrexia or high fever because the individual isn’t able ti give off eat through sweating.
Ultrastructural and morphometric studies of the peripheral nerves reveal loss of unmyelinated and small myelinated fibers and no innervation of sweat glands.
These features suggest that a defect in the differentiation and migration of neuronal crest elements and a possible deregulation of the NGF/NTRK1 pathway may be responsible for CIPA.
Many would feel that would be a great thin not to feel physical pain. Pain though, is a way one’s body tells it that something is wrong. An individual touching a hot stove would feel instant pain, forcing them to pull their hand away before causing too much demage. An individual with CIPA would keep their hand on the hot stove because they cannot feel in burning their hand. This is an important disease to study because by studing it,individuals can learn more about genetic diseases, how neurons and nerve work and communicate with the body and maybe find a cure for this disease.
CAUSE OF CONGENITAL INSENSIVITY TO PAIN WITH ANHIDROSIS
It has been reported recently that the NTRK1/NGF receptor gene is implicated in the pathogenesis of CIPA, this gene is responsible for encoding the receptor tyrosine kinase for nerve growth factor; point mutations within its tyrosine kinase (TK) domain have been detected in four patients with consanguinous parents.
A group of scientists have recently reported that one of those mutations, a GlyrArg substitution at codon 571, exerts a loss-of-function effect. The NTRK1 TK activity and biological effects are abolished when the Gly571Arg mutation is introduced into both the NTRK1 receptor and the constitutively active TRK-T3 oncogene.
Here we report the detection of a novel NTRK1 mutation associated with CIPA and demonstrate its inactivating effect. Nerve growth factor is critical for the formation of autonomic neurons and small sensory neurons in the dorsal root ganglia. TrkA is part of the TRK protooncogene family and is espressend in neaurons that sense temperature and noxious stimuli which is why a mutation to this gene causes the inability to feel pain. Mardy first reported that is a lack of innervation with eccrine sweart glands which affects the individual’s ability to sweat. CIPA is a very rare desease; there are only around 60 document cases in the United States and aroubnd 300 worldwide. It is hard though, to get the information on adults because many individuals do not live past the age of 25.
CLINICAL CASES
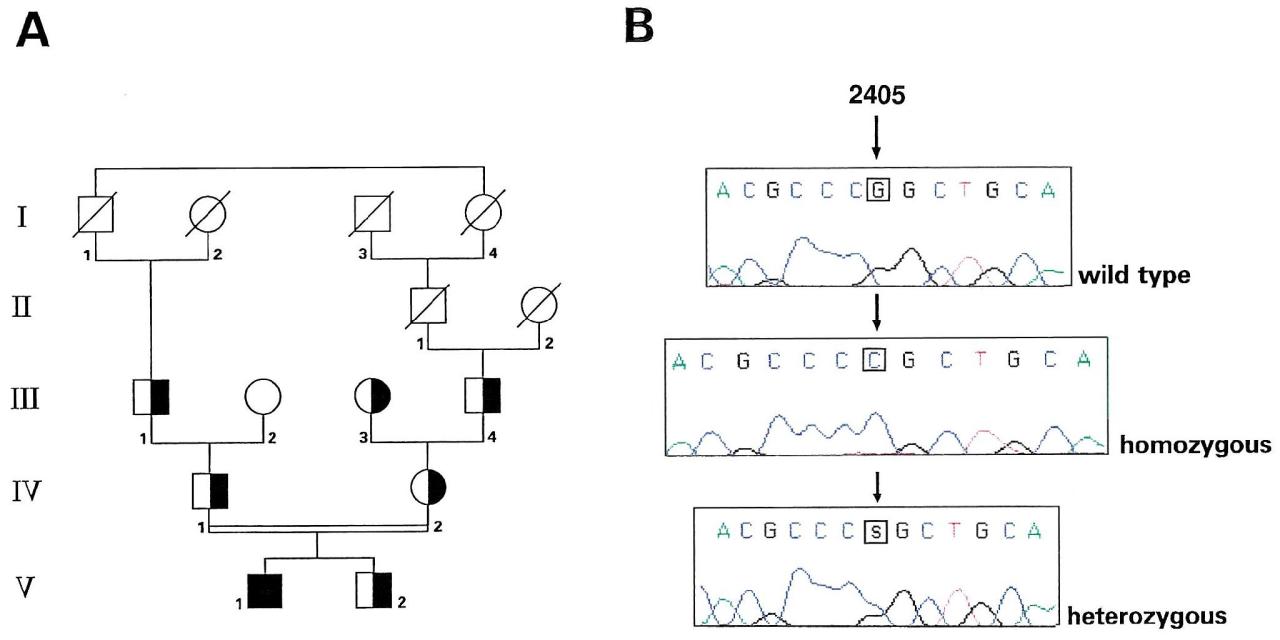
An Italian CIPA patient and his family (fig. 1) were screened for NTRK1 mutations. All the NTRK1 exons were amplified by PCR from peripheral blood lymphocite–DNA and sequenced by an ABI Prism 377 DNA sequencer. As a control, Hela DNA was amplified and sequenced simultaneously. A homozygous GrC transversion at nucleotide 2405 (exon 17, ArgrPro substitution at amino acid 774) was detected in the proband. All the other members of the family, including a younger, unaffected brother, are heterozygous, except for the paternal grandmother, who is homozygous for the wild-type sequence. Interestingly, the Arg774Pro mutation was present in both maternal grandparents, who had no documented consanguinity but were both from the same village.
To ascertain that the Arg774Pro mutation, rather than a rare polymorphism, was the cause of CIPA in the family, we performed functional studies, as reported for the Gly571Arg mutation, in an attempt to unveil a loss-of-function effect. The Arg774Pro mutation was introduced into the constitutively active TRK-T3 oncogene and the wild-type NTRK1 receptor cDNAs by sitedirected mutagenesis.
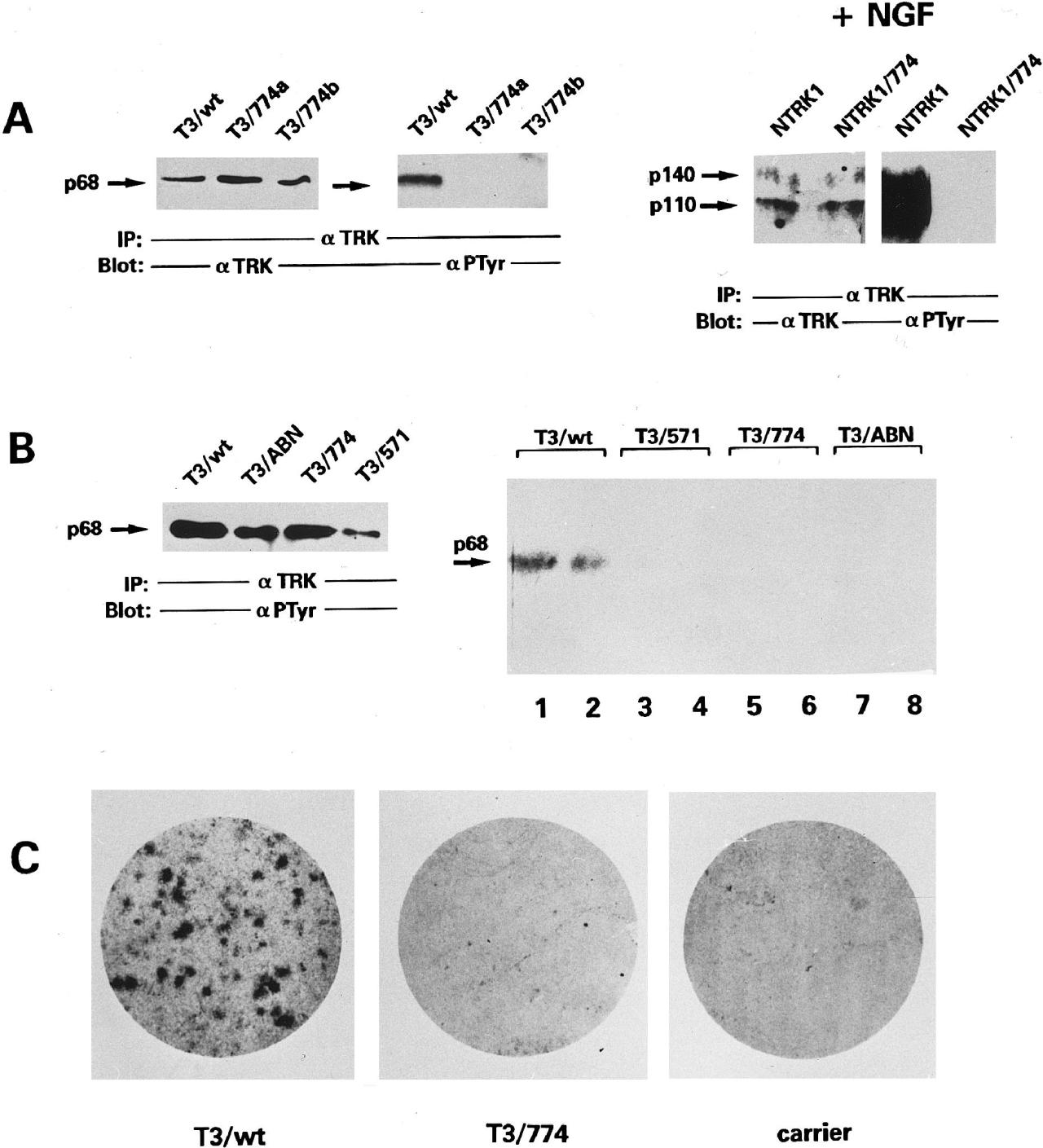
The mutated constructs (T3/774 and NTRK1/774) express protein of the expected molecular weight, as deduced by western blot analysis with anti-TRK antibodies (fig. 2A). Immunoblotting with antiphosphotirosine antibodies showed that the Arg774Pro mutation abrogates the constitutive phosphorylation of the TRK-T3 oncogene and the NGF-mediated phosphorylation of the wild-type NTRK1 receptor (fig. 2A). They analyzed the in vitro TK activity of mutant T3/774 with an immunocomplex autokinase assay. Two kinase-defective mutants (T3/ABN and T3/571) and the wild-type construct were used as negative and positive controls, respectively. As shown in figure 2B, the amounts of all the proteins were similar; the T3/774 and two kinase-defective mutants display an undetectable level of phosphorylation activity. In the NIH3T3 transfection/focus formation assay, no transformed foci arose from NIH3T3 cells transfected with T3/774 (fig. 2C), indicating that the introduced mutation abrogates the TRK-T3 transforming activity. All these findings demonstrate the loss-of-function effect of the Arg774Pro mutation, analogous to what was reported for the Gly571Arg mutation.
CONCLUSION
We have detected the following novel NTRK1 mutation associated with CIPA: a GrC trans version at nucleotide 2405 causing an ArgrPro substitution at codon 774. Biological and biochemical studies are consistent with a loss-of-function effect, demonstrating the pathogenic role of this mutation. Arg 774 is located at the C terminus of the NTRK1 TK domain; it is surrounded by residues conserved among the different members of TRK receptor family, but it is present only in NTRK1. Its role in receptor activation is unknown.
However, our data outline the importance of such residues for NTRK1 activity. It is interesting to note that All reported mutations associated with HSAN IV occur within the NTRK1 TK domain. Other point mutations occurring in the extracellular portion of NTRK1 or mutations of other genes encoding neurotrophin receptors might cause other variants of HSAN. Probably the most documentated case of CIPA was done on a Canadian woman named “Ms. C”. Her father was a physician and became aware of her disease when she didn’t respond to pain stimuli as a babt. When she was younger “Ms. C” received third- degree burns when she leanes on a radiator and also severed her tongue while chewing. She did not respond to electric shock, boiling water, ice baths, pinched tendons, or histamine injections. No physical changer in her body were observed and she reported no pain. She also reported that she couldn’t remember ever sneezing or coughing. This could be attributed to the fact tha coughing and sneezing are bodily functions which are caused by sensitivity and small pain to the throat and nose. Since she was unable to feel pain , her body did not respond to these stimuli either. Unfortunately, “Ms. C” died prematurely at the age of 29, frominfections in her knees, hips and spine. An interesting note though, “Ms. C” reported painful sensations during her last months of life. The cause of this phenomenon is still unexplained.
Patients must constantly be checked for scratches, bruises, bumps, anything that would indicate they are hurt. Before they eat, food must be checked to ensure it is not too hot. There are often dental and blood pressure problems which must constantly be monitorated. Whenever they wake up in the morning CIPA patients must check their eyes to ensure that they did not scratch their retina while they were sleeping. Individuals with CIPA often associate their lack of pain with immortality, which is why many patients die prematurely.
REFERENCES
First Reference
Second Reference
Vanessa Graziano \ Sonia Marchisio