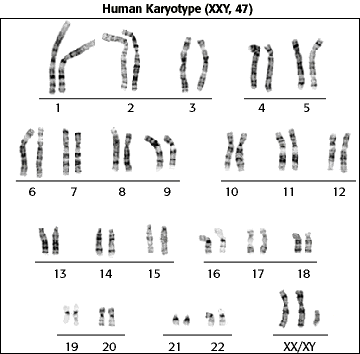
Klinefelter syndrome is the most common chromosomal aneuploidy in men (XXY karyotype, 1 in 600 live births) and results in testicular (infertility and androgen deficiency) and nontesticular (cognitive impairment and osteoporosis) deficits.
The extent to which skeletal changes are due to testosterone deficiency or arise directly from gene overdosage cannot be determined easily in humans. Klinefelter syndrome (KS) is the most frequent sex chromosome aneuploidy in human males.
KS is typified to date by reproductive dysfunction expressed as androgen deficiency, small testicular size, and infertility. Additional nonreproductive features such as tall stature and osteoporosis occur with early onset of or untreated hypogonadism from any cause.
On the other hand, certain behavioral, neurologic, psychiatric, and cognitive deficits are specific to KS and provide proof of principle that some features of KS cannot be mediated solely by androgen deficiency.
Epidemiology
XXY aneuploidy is the most common disorder of sex chromosomes in humans, with a prevalence of one in 500 males.
Other sex chromosomal aneuploidies are much less frequent with 48,XXYY and 48,XXXY being present in 1 per 17,000 to 1 per 50,000 male births.
The incidence of 49,XXXXY is 1 per 85,000 to 100,000 male births [Linden MG, Bender BG, Robinson A. Sex chromosome tetrasomy and pentasomy. Pediatrics.1995].
Cases of 46,XX males have also been reported.
Signs and symptoms
There are many variances within the XXY population, just as within the 46,XY population.
While it is possible to characterise XXY males with certain body types and physical characteristics, that in itself should not be the method of identification as to whether or not someone has XXY.
The only reliable method of identification is karyotype testing.
The degree to which XXY males are affected, both physically and developmentally, differs widely from person to person.
Cognitive and developmental
Some degree of language learning or reading impairment may be present, and neuropsychological testing often reveals deficits in executive functions, although these deficits can often be overcome through early intervention.
There may also be delays in motor development which can be addressed through occupational therapy and physical therapy.
XXY males may sit up, crawl, and walk later than other infants; they may also struggle in school, both academically and with sports.
Physical
As babies and children, XXY males may have weaker muscles and reduced strength.
As they grow older, they tend to become taller than average.
They may have less muscle control and coordination than other boys their age.
During puberty, the physical traits of the syndrome become more evident males may have larger breasts, weaker bones, and a lower energy level than other boys.
By adulthood, XXY males look similar to males without the condition, although they are often taller.
In adults, possible characteristics vary widely and include little to no signs of affectedness, a lanky, youthful build and facial appearance, or a rounded body type with some degree of gynecomastia (increased breast tissue).
XXY males are also more likely than other men to have certain health problems, which typically affect females, such as autoimmune disorders, breast cancer, venous thromboembolic disease, and osteoporosis.
In contrast to these potentially increased risks, it is currently thought that rare X-linked recessive conditions occur less frequently in XXY males than in normal XY males, since these conditions are transmitted by genes on the X chromosome, and people with two X chromosomes are typically only carriers rather than affected by these X-linked recessive conditions.
Affected males are often infertile, or may have reduced fertility.
Advanced reproductive assistance is sometimes possible.
Patients with Klinefelter syndrome undergo particular hormonal changes.
The serum follicle-stimulating hormone, luteinizing hormone, and inhibin B are at a normal level in prepubertal age, but they become abnormal over the time.
A study on adult individuals with the syndrome, found low testosterone levels in 45% of cases and 43.6% of patients accused sexual dysfunction.
Decreased testosterone levels are the result of testicular dysfunction with decrease in size of Leydig cells, and loss of germs and Sertoli cells leading to tubular hyalinization.
Increase in estradiol results from over-expression of aromatase CYP19. [ Wosnitzer MS, Paduch DA., Endocrinal issues and hormonal manipulation in children and men with Klinefelter syndrome. ].

Estradiol like other steroids, is derived from cholesterol. After side chain cleavage and using the delta-5 or the delta-4 pathway, androstenedione is the key intermediary. A fraction of the androstenedione is converted to testosterone, which in turn undergoes conversion to estradiol by an enzyme called aromatase. In an alternative pathway, androstenedione is aromatized to estrone, which is subsequently converted to estradiol.
Cause

Birth of a cell with karyotype XXY due to a non-disjunction event of one X chromosome from a Y
chromosome during meiosis I in the male.

Birth of a cell with karyotype XXY due to a non-disjunction event of one X chromosome during meiosis II in the female.
The extra X chromosome is retained because of a nondisjunction event during meiosis I (gametogenesis).
Nondisjunction occurs when homologous chromosomes, in this case the X and Y sex chromosomes, fail to separate, producing a sperm with an X and a Y chromosome.
Fertilizing a normal (X) egg produces an XXY offspring.
The XXY chromosome arrangement is one of the most common genetic variations from the XY karyotype, occurring in about 1 in 500 live male births.
Another mechanism for retaining the extra X chromosome is through a nondisjunction event during meiosis II in the female.
Nondisjunction will occur when sister chromatids on the sex chromosome, in this case an X and an X, fail to separate. (meiosis) An XX egg is produced which, when fertilized with a Y sperm, yields XXY offspring.
In mammals with more than one X chromosome, the genes on all but one X chromosome are not expressed; this is known as X inactivation.
This happens in XXY males as well as normal XX females.
However, in XXY males, a few genes located in thepseudoautosomal regions of their X chromosomes, have corresponding genes on their Y chromosome and are capable of being expressed.
The first published report of a man with a 47,XXY karyotype was by Patricia Jacobs and John Strong at Western General Hospital inEdinburgh, Scotland in 1959.
This karyotype was found in a 24-year-old man who had signs of Klinefelter syndrome.
Jacobs described her discovery of this first reported human or mammalian chromosome aneuploidy in her 1981 William Allan Memorial Award address.
Diagnosis

Percentages of Klinefelter's diagnosis divided by age groups, with most diagnoses occurring in adulthood.
About 10% of Klinefelter cases are found by prenatal diagnosis.
The first clinical features may appear in early childhoodor, more frequently, during puberty, such as lack of secondary sexual characters and aspermatogenesis, while tall stature as a symptom can be hard to diagnose during puberty.
Despite the presence of small testes, only a quarter of the affected males are recognized as having Klinefelter syndrome at puberty and 25% received their diagnosis in late adulthood: about 64% affected individuals are not recognized as such.
Often the diagnosis is made accidentally as a result of examinations and medical visits for reasons not linked to the condition.
The standard diagnostic method is the analysis of the chromosomes' karyotype on lymphocytes.
In the past, the observation of the Barr body was common practice as well.To confirm mosaicism, it is also possible to analyze the karyotype using dermal fibroblasts or testicular tissue.
Other methods may be: research of high serum levels of gonadotropins (follicle-stimulating hormone and luteinizing hormone), presence of azoospermia, determination of the sex chromatin,and prenatally via chorionic villus sampling oramniocentesis.
Differential diagnosis
The symptoms of Klinefelter syndrome are often variable; therefore, a karyotype analysis should be ordered when small testes, infertility, gynecomastia, long legs/arms, developmental delay, speech/language deficits, learning disabilities/academic issues and/or behavioral issues are present in an individual.
The differential diagnosis for the Klinefelter syndrome can include the following conditions: fragile X syndrome, Kallmann syndrome and Marfan syndrome. The cause of hypogonadism can be attributed to many other different medical conditions.
There have been some reports of individuals with Klinefelter syndrome who also have other chromosome abnormalities, such as Down syndrome.
Genetic counseling
The recurrence risk is not increased above that of the general population.
There is no evidence to suggest that a chromosomal nondisjunction process is likely to repeat itself in a particular family.
Antenatal diagnosis
Klinefelter syndrome can be detected prenatally by amniocentesis and cytogenetic amniotic fluid.
Parents should be counseled based on recent prospective and unbiased information.
Therapies
Decrease of aromatase
The techniques of assisted reproduction can give the possibility to have children of their own, but because the biopsy technique of sperm extraction (TESE) is invasive, it is always necessary to first evaluate the presence of a number of indicators that suggest the possible presence of residual spermatogenesis. The occurrence of spermatogonia and spermatozoa is more common in young people, for example, because the number of gametes decreases rapidly with age, but because of the increase in estradiol results from over-expression of aromatase CYP19, the administration of aromatase inhibitors and human chorionic gonadotrophin to stimulate the endogenous production of testicular testosterone , increases sperm count. The fractions of spermatozoa thus obtained demonstrate the presence of abnormal gametes, not euploid, 7-20% (in the male 46, XY, this share is less than 1%).
Testosterone treatment
Androgen replacement therapy should begin at puberty, around age 12 years, in increasing dosage sufficient to maintain age appropriate serum concentrations of testosterone, estradiol, FSH, and LH.
Androgen replacement promotes normalization of body proportions or development of normal secondary sex characteristics, but does not treat infertility, gynecomastia, and small testes.
Testosterone replacement also results in general improvement in behavior and work performance.
Testosterone also has beneficial long-term effects that might reduce the risk of osteoporosis, autoimmune disease, and breast cancer [Kocar IH, Yesilova Z, Ozata M, Turan M, Sengul A, Ozdemir I. The effect of testosterone replacement treatment on immunological features of patients with Klinefelter's syndrome. Clin Exp Immunol. 2000]
Speech therapy
Early identification and anticipatory guidance are important in boys with 47,XXY.
Early speech/language therapy is particularly essential in helping the child to develop skills in the understanding and production of more complex language.
Physical therapy
Physical therapy should be considered for boys who have hypotonia or delayed in gross motor skills which may impact the muscle tone, balance, and coordination.
Occupational therapy
If boys with 47,XXY have fine motor dyspraxia, occupational therapy should be recommended. In addition, an occupational therapist may benefit infants with 47,XXY who have feeding problems or difficulty with latching on or sucking.
Educational services
Males with 47,XXY should receive a comprehensive psychoeducational evaluation to assess their areas of strengths and weaknesses. The information obtain from this evaluation may be helpful in planning appropriate resources and classroom placement. Consultation with a developmental-behavioral pediatrician is suggested....