HYPOXIA AS AN OBSTACLE IN PULMONARY SURFACTANT PRODUCTION
Pulmonary surfactant is a surface-active lipoprotein complex (phospholipoprotein) secreted by type II alveolar cells. The proteins and lipids that the surfactant comprises have both a hydrophilic and a hydrophobic region. By apposing to the air-water interface of alveoli, with the hydrophilic head groups by the water and the hydrophobic tails facing towards the air, the main lipid component of surfactant, dipalmitoylphosphatidylcholine (DPPC), reduces surface tension. It's mainly composed by dipalmitoylphosphatidylcholine (DPPC) and other phospholipids. DPPC is a phopsholipid with two 16-carbon saturated chains and a phosphate group with quaternary amine group attached. The DPPC is the strongest surfactant molecule in the pulmonary surfactant mixture. It also has higher compaction capacity than the other phospholipids, because the apolar tail is less bent. Nevertheless, without the other substances of the pulmonary surfactant mixture, the DPPC's adsorption kinetics is very slow. This happens primarily because the phase transition temperature between get to liquid crystal of pure DPPC is 41°C, which is higher than the human body's. DPPtdCho is synthesized mainly through remodeling of phosphatidilcoline.
It is thought that a lysophosphatidylcholine (lysoPC) acyltransferase may play a critical role in its synthesis. The identity of this acyltransferase has not yet been confirmed.Dipalmitoylphosphatidylcholine is an exception to the rule of thumb that biological phospholipids are synthesized with a saturated fat at the R1 position and an unsaturated fat at the R2 position.
Phosphatidylcholine molecules form ~85% of the lipid in surfactant and have saturated acyl chains. Phosphatidylglycerol (PG) forms about 11% of the lipids in the surfactant, it has unsaturated fatty acid chains that fluidize the lipid monolayer at the interface. Neutral lipids and cholesterol are also present. The components for these lipids diffuse from the blood into type II alveolar cells where they are assembled and packaged for secretion into secretory organelles called lamellar bodies.
Pulmonary surfactant is as well important for the correct operation of the respiratory system as it carries on several functions such as:
- Increasing pulmonary compliance.
- Preventing atelectasis (collapse of the lungs) at the end of expiration.
- Easing recruitment of collapsed airways.

The production of pulmonary surfactant can be alterated by the presence of hypoxia (that is briefly a condition in which the body or a region of the body is deprived of adequate oxygen supply), especially if this happens during fetal life, by acting directly on the type II alveolar cells. In fact, experiments conducted on fetal rat lung, show how a severe hypoxia can be related with several problems of those cells, such as slowed cell growth, apparently due to the block of S phase cell progression into G2 and/or by activating S phase transit into apoptosis (in fact hypoxia induces epithelial cell apoptosis), which can be avoided simply with a 24h recovery in normoxia (21% O2). It's important to specify that a period of normoxic recovery reversed the hypoxia-induced alterations in cell proliferation and apoptosis but did not completely reverse effects on epithelial cell function. Hypoxia, in fact, induces both necrosis and apoptosis, and the proportion of these two modes is highly dependent on the cell type. Overexpression of Bcl-2 or Bcl-X, blocks hypoxia-induced apoptosis in a dose-dependent manner. Summing up, hypoxia transiently increases expression of p53 and of Mch3/ICE-LAP6, but not of other ICE/Ced-3-like apoptotic cysteine proteases. The G2 cell cycle block appeared to be driven by hypoxic dose-dependent increases in the CDK inhibitors, p27 and p21. We conclude that hypoxia decreases pulmonary surfactant production, blocks epithelial cell proliferation and activates alveolar type II cells apoptosis.
Moreover, exposure to hypoxia impairs alveolar edema clearance by downregulation of both ENaC and the Na/K-ATPase function.

Induction of Apoptosis as well as Necrosis by Hypoxia and Predominant Prevention of Apoptosis by Bcl-2 and Bcl-X, 1996.
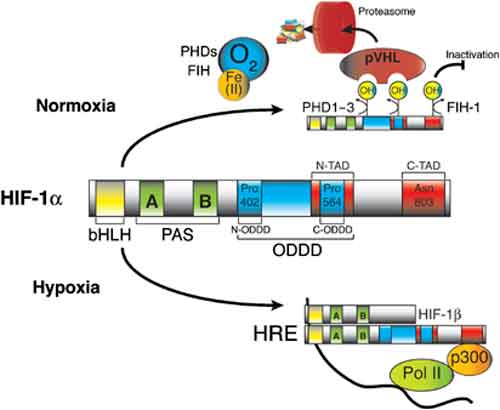
Other experiments analyzed the reduction of the pulmonary surfactant production by inquiring the role that hypoxia-inducible factors had in it. It seems that the air-liquid interface condition maintained the expression of surfactant proteins, whereas the expression was down-regulated under the submerged condition, and the effect was rapid and reversible. Under submerged conditions, there was an increase in HIF1α and HIF2α in nuclear extracts, mRNA levels of HIF inducible genes, vascular endothelial growth factor, glucose transporter-1 (GLUT1), and a greater amount of protein pyruvate dehydrogenase kinase lisozyme-1. The expression of surfactant proteins was suppressed and GLUT1 mRNA levels were induced when cells were cultured with 1 mM dimethyloxalyl glycine. The expression of surfactant proteins was restored under submerged conditions with supplemented 60% oxygen.
Moreover, HIF-1 is thought to be involved in apoptotic and anti-apoptotic processes. In fact severe or prolonged hypoxia rather induces apoptosis: that is, at least in part, initiated by the direct association of HIF-1α, p53 and p53-induced gene expression. On the other hand, when HIF-1α dimerized with ARNT, as an active transcription factor, il could protect cells from apoptosis induced by several conditions.
Hypoxia can also decrease the surfactant protein expression in which, in this regard, also ipercapnia has an important role. In fact alveolar epithelial cells produce surfactant proteins, including surfactant protein C, when cultured under normal conditions, which are reduced under hypoxic conditions. Specifically, pro-SpC expression is moderately decreased after 8h of culture in hypoxia, and is completely attenuated after 48h. Hypercapnia decreases pro-SpC expression only after 48h of exposure. As previously said, hypoxic culture of alveolar cells results in progressive arrest of cells in G1 phase of the cell cycle and increases apoptosis, which is first observed 4h after the exposure and peaks after 24h. In contrast hypercapnia has no significant effect on alveolar epithelial cell proliferation or apoptosis.
From these experiments, we can assume that hypoxia can directly decrease the production of pulmonary surfactant inducing apoptosis by itself, by reducing the expression of surfactant proteins or by inducing HIFs that, in some cases, can contribute to the beginning of apoptosis.

But hypoxia can also acts in a indirect way to decrease the production of pulmonary surfactant. In fact hypoxia and HIFs can decrease the production of thyroid hormones T3 and T4.
Briefly, thyroid hormones triiodothyronine (T3) and thyroxine (T4) are tyrosine-based hormones produced by the thyroid gland that are primarily responsible for the regulation of metabolism. Iodine is necessary for the production of T3and T4. A deficiency of iodine leads to decreased production of T3 and T4, enlarges the thyroid tissue and causes a disease known as goitre. The major form of thyroid hormone in the blood is thyroxine (T4), which has a longer half-life than T3.The ratio of T4 to T3 released into the blood is roughly 20 to 1. T4 is converted into the active T3 (three to four times more powerful than T4) within cells by deiodinase.

Hypoxia and HIFs exert their effects right on deiodinase enzyme, especially in presence of hypoxic-ischaemic disease Hypoxia-inducible factor induces local thyroid hormone inactivation during hypoxic-ischemic disease in rats.. 2008. In fact hypoxia induces the expression of D3 gene, DIO3, by an hypoxia-inducible factor-dependent (HIF-dependent) pathway. D3 activity and mRNA production were increased both by hypoxia and by hypoxia mimetics that increase HIF-1. HIF-1α interacted specifically with the DIO3 promoter, indicating that DIO3 may be a direct transcriptional target of HIF-1. Using a rat model of cardiac failure due to RV hypertrophy, it was found that HIF-1α and D3 proteins were induced specifically in the hypertrophic myocardium of the RV, creating an anatomically specific reduction in local T3 content and action. These results suggest a mechanism of metabolic regulation during hypoxic-ischemic damage in which HIF-1 reduces local thyroid hormone signaling through induction of D3.

In fact, an experiment on diabetic and hypophysectomized rats, compared to normal ones, shows how the administration of triiodothyronine (T3), to the diabetic and hypophysectomized rats restored the normal activities of palmitoyl-CoA synthetase, phospholipase A2, and lysophosphatidylcholine acyltransferase enzymes. So the administration of T3 stimulated the rate of synthesis of this enzyme indicating increasing synthesis of this enzyme and not of activation of the pre-existing inactive species. Reduced phospholipid contents, specially decreased amount of lecithin and dipalmitoyl lecithin (DPL) were observed in the lungs of the diabetic and hypophysectomized animals as compared to those in the normal animals. T3 also increased the lecithin and DPL content of the normal rat lungs. These results provide evidence for the involvement of the thyroid hormones in the control of the pulmonary surfactant. In presence of hypoxa, because of a decreased production of T3 and T4, all these events can't happen.