Alessio Vitale, Francesca Pegoraro
DEFINITION
Stevens–Johnson Syndrome is named from Albert Mason Stevens and Frank Chambliss Johnson, American pediatricians who jointly published a description of the disorder in the American Journal of Diseases of Children in 1922. Stevens–Johnson syndrome (SJS) and toxic epidermal necrolysis (TEN) are two forms of a life-threatening skin conditions, in which apoptosis causes the epidermis to separate from the dermis.
Pubmed, 'Oral lesions associated with Nevirapine-induced Stevens-Johnson syndrome and toxic epidermal necrolysis: A report of 10 cases', 2013

Stevens-Johnson Syndrome is a form of immune-complex–mediated hypersensitivity that typically involves the skin and the mucous membranes, characterized by severe erythema, multiform-like eruption of the skin, lesions of the oral, genital and anal mucosa, hemorrhagic crusting on the lips, associated with fever, headache and arthralgia.
The simplest classification breaks the disease down as follows, distinguishing the type of lesions and the amount of body surface interested:
- Stevens-Johnson syndrome: a minor form of toxic epidermal necrolysis, with less than 10% body surface area ( BSA ) detachment
- Overlapping Stevens-Johnson syndrome ( overlapping TEN ): detachment of 10-30% of the BSA
- Toxic epidermal necrolysis ( TEN ): Detachment of more than 30% of the BSA
EPIDEMIOLOGY
These conditions were first recognised in 1922.
Since then, the overlap has been stated to be of 2-3 cases per 1 million population per year.
For SJS and TEN the distribution of gender is almost equal (even though slightly more females).
The mortality is almost 10% for patients with SJS, approximately 30% for patients with SJS/overlapping TEN and almost 50% for patients with TEN.
Severe drug hypersensitity reactions as Stevens-Johnson syndrome and toxic epidermal necrolysis develop MORE OFTEN in HIV-infected patients compared to other populations. In fact, multiple drugs, that are usually prescribed for prevention or treatment of opportunistic infections and antiretroviral, pose these patients on a higher risk of developing drug hypersensitivity, as well as antiretroviral agents.
Pubmed, 'Drug hypersensitivity in human immunodeficiency virus-infected patient: challenging diagnosis and management', 2014
In Europe, approximately 5% of the patients with SJS/TEN were HIV-infected, but the number seems to have decreased in the past decade. As expected, the distribution of age and gender differs between HIV-infected and non-HIV-infected patients with SJS/TEN, while mortality rate and outcome are comparable.
SYMPTOMS
Stevens–Johnson syndrome (SJS) usually begins with fever, sore throat, and fatigue, which is commonly misdiagnosed and therefore treated with antibiotics.
Ulcers and other lesions begin to appear on the mucous membranes, almost always in the mouth and lips but also in the genital and anal regions. Those in the mouth are usually extremely painful and reduce the patient's ability to eat or drink.
Other typical prodromal symptoms of Stevens-Johnson syndrome
are as follows:
- Cough productive of a thick, purulent sputum
- Headache
- Malaise
- Arthralgia
Patients may complain of a burning rash that begins symmetrically on the face and the upper part of the torso. The cutaneous lesions are characterized as follows:
- The rash can begin as macules that develop into papules and finally into confluent erythema

- The typical lesion has the appearance of a target; this is considered pathognomonic
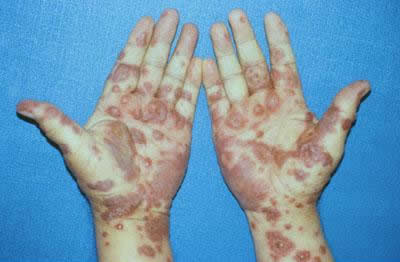
- In contrast to the typical lesions of erythema multiforme, these lesions have only 2 zones of color
- Lesions may become bullous and later rupture, leaving denuded skin; the skin becomes susceptible to secondary infection
- Urticarial lesions typically are not pruritic
- Infection may be responsible for the scarring associated with morbidity
- The rash may be confined to every area of the body, most often the trunk
Signs of mucosal involvement can include erythema, blistering, ulceration and necrosis.
The following ocular signs may be noted on slit-lamp examination:
- Eyelids: Trichiasis, distichiasis, meibomian gland dysfunction, blepharitis
- Conjunctiva: Papillae, follicles, keratinization, subepithelial fibrosis, conjunctival shrinkage, foreshortening of fornices, symblepharon, ankyloblepharon
- Cornea: Superficial punctate keratitis, epithelial defect, stromal ulcer, neovascularization, keratinization, limbitis, conjunctivalization, stromal opacity, perforation

DIAGNOSIS
Diagnosis is based on clinical classification and on histopathology.
Minimal dermal inflammatory cell infiltrate and full-thickness necrosis of the epidermis are typical histopathologic findings in patients with Stevens-Johnson syndrome. Histopathologic examination of the skin can also reveal the following:
- Changes in the epidermal-dermal junction ranging from vacuolar alteration to subepidermal blisters
- Dermal infiltrate: superficial and mostly perivascular
- Apoptosis of keratinocytes
- The dermoepidermal junction and epidermis is infiltrated mostly by CD8+ T lymphocytes
ETIOLOGY
Stevens–Johnson syndrome (SJS) is thought to arise from a disorder of the immune system (it is a type of hypersensitivity reaction). The immune reaction can be triggered by drugs or infections (less frequently): Approximately 75% of SJS/TEN are caused by medications, and 25% by infections and 'other' causes. Genetic factors are associated with a predisposition to SJS (see later).
Medications that can cause this reaction include:
- Barbiturates (sedatives)
- Penicillins (antibiotics)
- Phenytoin (antiepileptic)
- Sulfonamides (antibiotics)
- Non-steroidal anti-inflammatory drugs
Roughly 100-200 different drugs may be associated with SJS.
Infections include:
- Herpes simplex
- Mycoplasma
- Epstein-Barr virus
- Diphtheria,
- Mycobacteria
Pubmed, 'A European study of HLA-B in Stevens-Johnson syndrome and toxic epidermal necrolysis related to five high-risk drugs', 2008
PROPOSED PATHOGENESIS MECHANISM
As explained previously, drugs are the etiologic factor in the majority of SJS/TEN cases. However, the molecular mechanisms involved in the pathogenesis of toxic epidermal necrolysis (TEN) remain not fully understood.
Drug specific CD8+ cytotoxic lymphocytes can be detected in the early blister fluid. They have natural killer cell activity and can kill keratinocytes by direct contact. Prior in vitro studies suggest that a MHC class I restricted-drug presentation leads to a clonal expansion of CD8+CTLs, and induces immune reactions. The MHC-restricted presentation of a drug or its metabolite for T-cell activation is supported by recent findings of strong genetic association between HLA-B alleles and reactions to specific drugs.
A genetic predisposition for SJS/TEN has long been discussed. It had been suggested an association with certain HLA types more than 20 years ago, demonstrating that 100% of Han-Chinese patients with SJS/TEN due to the use of carbamazepine were positive for the allele HLA-B 1502. This finding could not be confirmed in Europe showing that ethnicity matters more than previously thought in this context.
For allopurinol-induced cases of SJS/TEN a 100% association with HLA-B 5801 could be demonstrated in a Han-Chinese population, whereas in the European population the association was present in no more than 55%.
Strong associations such as those in Han-Chinese suggest that these alleles must be involved in the presentation of a specific drug antigen in a better way than other HLA alleles. Thus, the risk of SJS/TEN is not only related to the exposure with high-risk drugs, but also to a genetic predisposition.
There are probably two major pathways involved in the pathogenesis:
- Fas-Fas ligand pathway of apoptosis, considered a pivotal step in the pathogenesis of TEN. The Fas ligand (FasL), a form of tumor necrosis factor, is secreted by blood lymphocytes and can bind the Fas ‘death’ receptor expressed by keratinocytes.
- T cells, especially CD8+ lymphocytes, have been identified to play an important role in the process, through the releasing of granzymes and granulisin.
TISSUE DESTRUCTION MEDIATED BY FAS IN SJS
It has been recently shown that SJS is associated with highly increased keratinocyte FasL expression, together with conserved levels of keratinocyte Fas expression in vivo.
It is well known that primary keratinocytes are sensitive to the cytolytic effect of FasL in vitro, and this sensitivity can be further enhanced by interferon gamma, a cytokine known to be present in the skin during TEN. However, it is still not fully understood what causes the up-regulation of FasL/Fas on keratinocytes, and how the immune system, including T cells found in blister fluid at the onset of disease may regulate this.
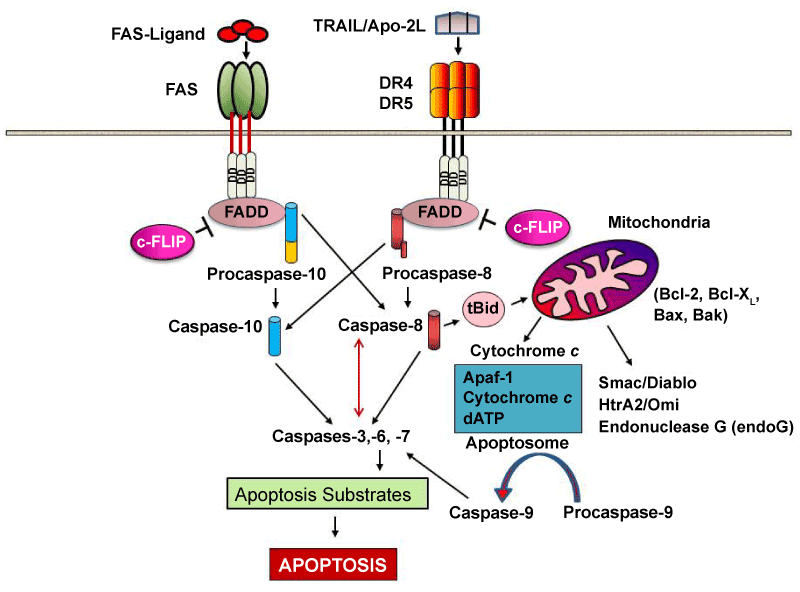
Cells are equipped with surface sensors named Death receptors. They are a growing family of transmembrane proteins which can detect the presence of specific extracellular death signals and rapidly trigger cellular destruction by apoptosis. Human death receptors (Fas, TNF-R1, TRAMP, TRAIL-R1, TRAIL-R2 and DR-6) have been identified. They all contain 2–4 cystein-rich extracellular domains and a cytoplasmic sequence named “ death domain ” (DD). The best studied to date is Fas (CD95) and its ligand (FasL, CD95L).
Recently, strong evidence has shown that deregulation of Fas expression and/or signalling contributes to the pathogenesis of toxic epidermal necrolysis. With these new developments, strategies for modulating the function of Fas signalling have emerged and opened up novel therapeutic possibilities. Specific blockade of Fas, for example with intravenous immunoglobulin preparations containing specific anti-Fas antibodies, has shown great promise in the treatment of toxic epidermal necrolysis. However the actual patient treatment is limited to intensive supportive care.
The signaling implies multimerisation of Fas upon binding of the membrane- bound form of FasL recruits the bipartite adaptor molecule FADD (Fas-associated death domain; composed of an amino terminal death effector domain [DED] and a carboxyl terminal DD). FADD binds to Fas (via hemophilic DD–DD interactions) and recruits the DED-containing caspase-8 (or probably also caspase-10) to the receptor via hemophilic DED–DED interactions. Caspase-8 (or -10) within this newly formed death-inducing signaling complex ( DISC ) then proteolytically autoactivates itself and initiates apoptosis by subsequent cleavage of downstream effector caspases (caspases-3, -6, -7) leading to DNA degradation, membrane blebbing, and other hallmarks of apoptosis.
In most cell types, caspase-8 catalyzes the cleavage of the pro-apoptotic protein Bid into its truncated form, tBid. tBid engages exclusively anti-apoptotic proteins (Bcl-2, Bcl-xL), allowing Bak and Bax to translocate to the outer mitochondrial membrane, thus permeabilizing it and facilitating release of pro-apoptotic proteins such as cytochrome c and Smac/DIABLO, an antagonist of inhibitors of apoptosis proteins (IAPs).
ROLE OF GRANZYMES AND GRANULYSIN
Granule-mediated exocytosis via perforin and granzyme B results in cell death. Perforin and granzyme B can be detected in early blister fluid and it has been suggested that high levels may be associated with disease severity. CD8+ T cells from the blister fluid of patients with TEN induced by drugs were tested for their cytotoxic function and reacted without restimulation against the parent drug, but not against the metabolite. This finding challenged the hypothesis that metabolites may be directly involved in the process of epidermal cell death. T-cell activation by drug antigens requires the interaction of the T-cell receptor (TCR) with the MHC on antigen-presenting cells. Thus, the drug may bind to the MHC molecule, which is recognized by the TCR leading to specific TCR activation, or the drug may bind first to a specific TCR that then interacts with the MHC. Both ways are possible, but drugs with a strong association to specific HLA alleles (as seen above) are more suggestive to interact primarily with the HLA molecule.
GRANZYMES: Granzymes are serine-proteases that are released by cytoplasmic granules within cytotoxic T cells and natural killer cells. Their purpose is to induce apoptosis within infected or damaged cells, thus destroying them. The goal of the granules and perforins is to create a pathway for the granzymes to follow and enter the target cells cytosol. In fact cytotoxic T cells and natural killer cells release this protein, which attacks the target cells.

A multimeric complex (Granzyme B, perforin, and granulysin) can enter a cell through the mannose 6-phosphate receptor and is enclosed in a vesicle. Perforin then allows Granzyme B to pass through the vesicle surface and into the cell, causing apoptosis by various pathways. They do so by cleaving caspases (especially caspase-3), which in turn activate caspase-activated DNase. Also, Granzyme B cleaves the protein Bid, which recruits the protein Bax and Bak to change the membrane permeability of the mitochondria, causing the release of cytochrome c (which is one of the parts needed to activate caspase-9 via the Apoptosome), Smac/Diablo (which suppress the inhibitor of apoptosis proteins(IAPs)), among other proteins. As well, Granzyme B is shown to cleave many of the chemicals responsible for apoptosis without the aid of caspase.

GRANULYSIN: Granulysin is an antimicrobial and cytotoxic molecule expressed in granules of CTL and NK cells. It has been shown that granulysin damages cell, and causes release of cytochrome c. Granulysin-induced apoptosis is blocked in cells overexpressing Bcl-2. Activation of caspase 3 is observed in granulysin-treated cells, suggesting that granulysin activates a novel pathway of CTL- and NK cell-mediated death distinct from granzyme induced apoptosis.
The 15-kDa granulysin is secreted by CTLs and NK cells and is can be found in blister fluid in SJS/TEN. This has a cytotoxic effect on keratinocytes and also acts as a chemoattractant for T cells, monocytes and other inflammatory cells. Granulysin concentrations in the blister fluids were two to four times higher than perforin, granzyme B or soluble Fas ligand concentrations, and depleting granulysin reduced the cytotoxicity. Granulysin in the blister fluids was a 15-kDa secretory form, and injection of it into mouse skin resulted in features mimicking SJS-TEN. It has been demonstrated that secretory granulysin is a key molecule responsible for the disseminated keratinocyte death in SJS-TEN.
Pubmed, 'A distinct pathway of cell-mediated apoptosis initiated by granulysin', 2001
INTRAVASCULAR COAGULATION HYPOTESIS
In SJS syndrome the involvment of intravascular coagulation is possible; however, medical literature lacks information about the topic.
Despite that, we can theorize that massive cytokine release from activated leucocytes drives the beginning of coagulation. Supporting this there are some articles, showing an association between cytokine release and coagulation. The subsequent ischaemia could worsen the organ epithelial damage due to CD8+ attack.
In fact, T-cell activation results in expression of cytokines including TNF-α, interferon-γ, granzyme B and granulysin. Besides their role in causing SJS/TEN, these cytokines cause coagulation activation: the release of these cytokines causes immune-mediated activation of coagulation with increase in expression of TAT, MCP-1, F1.2 (protein fragment released by Prothrombin's cleavage by factor X) and platelet microparticles and corresponding decrease of protein C (that inactives proteins Factor Va and Factor VIIIa), antithrombin, and PAI-1. These alterations in coagulation may progress to coagulopathy and disseminated intravascular coagulation.
Immune-Mediated Activation Of Coagulation In Patients With Stevens Johnson Syndrome
COMPLICATIONS
SJS can be fatal due to complications in the acute phase that include:
- Dehydration and acute malnutrition
- Infection of skin, mucous membranes, lungs; septicemia
- Shock and multiple organ failure, including kidney failure
- Thromboembolism and disseminated intravascular coagulopathy
Longterm complications include:
- Pigment change – patchwork of increased and decreased pigmentation
- Skin scarring, especially at sites of pressure or infection
- Loss of nails with permanent scarring (pterygium) and failure to regrow
- Scarred genitalia
- Serious eye problems, which can lead to blindness. This is the most important of the longterm complications. These include:
- Dry and/or watery eyes, which may burn and sting when exposed to light
- Conjunctivitis: red, crusted, or ulcerated conjunctiva
- Corneal ulcers, opacities and scarring
- Symblepharon: adhesion of conjunctiva of eyelid to eyeball

- Ectropion or entropion: turned-out or turned-in eyelid
- Trichiasis: inverted eyelashes
- Synechiae: iris sticks to cornea
THERAPY
Care of a patient with SJS/TEN requires:
- Cessation of suspected causative drug(s)
- Nutritional and fluid replacement by intravenous and nasogastric routes
- Temperature maintenance – as body temperature regulation is impaired
- Pain relief – as pain can be extreme
- Skin care (topical antiseptics,…)
- Eye care: (antiseptic and antibiotic eye drops)
- Urinary catheter because of genital involvement and immobility