DEFINITION
Hirschsprung disease ( HSCR) also called congenital aganglionic megacolon,is a disorder of the abdomen that occurs when part or all of the large intestine or antecedent parts of the gastrointestinal tract have no nerves and therefore cannot function. The major gene of Hirschsprung disease was identified in this chromosomal 10 region, it was the RET proto-oncogene .
INTRODUCTION
The first report of Hirschsprung disease dates back to 1691, however, the disease is named after Harald Hirschsprung, the Danish physician who first described two infants who died of this disorder in 1888.
Hirschsprung’s disease is a congenital disorder of the colon in which certain nerve cells, known as ganglion cells, are absent, causing chronic constipation. The lack of ganglion cells is in the myenteric plexus , which is responsible for moving food in the intestine.
During normal fetal development, cells from the neural crest migrate into the large intestine (colon) to form the networks of nerves called Auerbach's plexus and Meissner's plexus. In Hirschsprung's disease, the migration is not complete and part of the colon lacks these nerve bodies that regulate the activity of the colon. The affected segment of the colon cannot relax and pass stool through the colon, creating an obstruction.People with HSCR are born with it and are usually diagnosed when they are infants. Less severe cases are sometimes diagnosed when a child is older. An HD diagnosis in an adult is rare. (Hirschsprung disease)

- There are four types of Hirschsprung disease:
- In 80% of individuals, aganglionosis is restricted to the rectosigmoid colon ( short-segment disease).
- In approximately 15%-20%, the aganglionosis extends proximal to the sigmoid colon ( long-segment disease).
- In approximately 5% of individuals, aganglionosis affects the entire large intestine ( total colonic aganglionosis).
- Rarely, the aganglionosis extends into the small bowel or even more proximally to encompass the entire bowel ( total intestinal aganglionosis)

The incidence of short-segment disease (80% of HSCR) is four times greater in males than in females; equal numbers of males and females present with long-segment HSCR.
(A genetic study of Hirschsprung disease,1990)
(Hirschsprung Disease Overview,2011)
EPIDEMIOLOGY
The incidence of HSCR is approximately 1 in 5000 live births
The incidence varies among different ethnic groups:
- Persons of northern European origin: 1.5 in 10,000 live births
- African Americans: 2.1 in 10,000
- Asians: 2.8 in 10,000
HSCR occurs as an isolated trait in 70% of patients, is associated with chromosomal anomaly in 12% of cases, and occurs with additional congenital anomalies in 18% of cases.
HSCR is a multifactorial, non-mendelian disorder in which rare high-penetrance coding sequence mutations in the receptor tyrosine kinase RET contribute to risk in combination with mutations at other genes.
Hirschsprung's disease causes about 25% of all newborn intestinal blockages. It occurs five times more often in males than in females. Sometimes is associated with other inherited or congenital conditions, such as Down syndrome (10% of cases).
( Hirschsprung disease, associated syndromes, and genetics: a review, 2008 )
(HIRSCHSPRUNG DISEASE, SUSCEPTIBILITY TO, 1; HSCR1,2012)
(A common sex-dependent mutation in a RET enhancer underlies Hirschsprung disease risk,2005)
SYMPTOMS
The symptoms of HSCR are constipation or intestinal obstruction, usually appearing shortly after birth. Constipation in infants and children is common and usually comes and goes, but if your child has had ongoing constipation since birth, HSCR may be the problem.
Symptoms in Newborns

Newborns with HSCR almost always fail to have their first bowel movement within 48 hours after birth. Other symptoms include:
- Green or brown vomit
- Explosive stools after a doctor inserts a finger into the rectum
- Swelling of the belly, also known as the abdomen
- Lots of gas
- Bloody diarrhea
Symptoms in Symptoms in Toddlers and Older Children
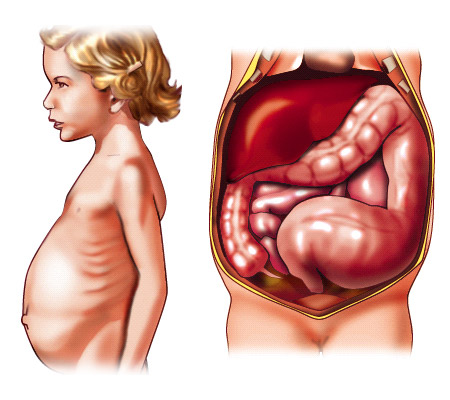
Symptoms of HSCR in toddlers and older children include:
- Not being able to pass stools without laxatives or enemas. A laxative is medicine that loosens stool and increases bowel movements. An enema is performed by flushing water, or sometimes a mild soap solution, into the anus using a special wash bottle.
- Swelling of the abdomen.
- Lots of gas.
- Bloody diarrhea.
- Slow growth or development.
- Lack of energy because of a shortage of red blood cells, called anemia.
( What I need to know about Hirschsprung Disease,2010 )
INHERITANCE
HSCR is considered to be a polygenic disorder with incomplete penetrance, variable expressivity, and a 4:1 predominance in males.
Approximately 20 percent of cases occur in multiple members of the same family. The remainder of cases occur in people with no history of the disorder in their families.
Hirschsprung disease apears to have a dominant pattern of inheritance, which means one copy of the altered gene in each cell may be sufficient to cause the disorder. The inheritance is considered to have incomplete penetrance because not everyone who inherits the altered gene from a parent develops Hirschsprung disease.
(Hirschsprung Disease Overview,2011 )
(Hirschsprung disease,2012)
PATHOGENESIS
The most accepted theory of the cause of Hirschsprung is that there is a defect in the craniocaudal migration of neuroblasts originating from the neural crest that occurs during the first 12 weeks of gestation.
Lipson in 1989, raised the question of hyperthermia in early gestation as a factor in Hirschsprung disease.
After,Larsson could not confirm a correlation between hyperthermia during pregnancy and Hirschsprung disease in the offspring.
(Hirschsprung disease in the offspring of mothers exposed to hyperthermia during pregnancy,1989 )
(No correlation between hyperthermia during pregnancy and Hirschsprung disease in the offspring,1989)
CAUSES
Isolated Hirschsprung disease can result from mutations in one of several genes, including the RET, EDNRB, and EDN3 genes.
Chromosomal Causes
A chromosomal abnormality is present in approximately 12% of individuals with HSCR.
Several genes and specific regions on chromosomes (loci) have been shown or suggested to be associated with Hirschsprung's disease:
- RET
Mutations in the RET gene (on chromosome 10) are the most common known genetic cause of Hirschsprung disease. The RET gene provides instructions for producing a protein that is involved in signaling within cells. This protein appears to be essential for the normal development of several kinds of nerve cells, including nerves in the intestine. Mutations in the RET gene result in a nonfunctional version of the RET protein that cannot transmit signals within cells. Without RET protein signaling, enteric nerves do not develop properly. Absence of these nerves leads to the intestinal problems characteristic of Hirschsprung disease.
Mutations in RET appear to be dominant loss-of-function mutations with incomplete penetrance(50%-70%) and variable expressivity.
(RET,2013 )




- EDNRB
The EDNRB gene (chromosome 13) provides instructions for making a protein called endothelin receptor type B. This protein is located on the surface of cells and functions as a signaling mechanism, transmitting information from outside the cell to inside the cell. The receptor interacts with proteins called endothelins to regulate several critical biological processes, including the development and function of blood vessels, the production of certain hormones, and the stimulation of cell growth and division.
(EDNRB,2012)


- GDNF
The GNDF gene (chromosome 5) encodes a highly conserved neurotrophic factor. The recombinant form of this protein was shown to promote the survival and differentiation of dopaminergic neurons in culture, and was able to prevent apoptosis of motor neurons induced by axotomy. The encoded protein is processed to a mature secreted form that exists as a homodimer. The mature form of the protein is a ligand for the product of the RET (rearranged during transfection) protooncogene. Mutations in this gene may be associated with Hirschsprung disease-3.
(GNDF,2014 )


- EDN3
The EDN3 gene (chromosome 20) provides instructions for making a protein called endothelin 3.
Endothelin 3 is one of the proteins that interacts with endothelin receptor type B. During early development before birth (embryonic development), endothelin 3 and endothelin receptor type B together play an important role in neural crest cells. These cells migrate from the developing spinal cord to specific regions in the embryo, where they give rise to many different types of cells. In particular, endotelin 3 and its receptor are essential for the normal formation of nerves in the intestine (enteric nerves) and melanocytes.
(EDN3,2012)


Associated syndromes
The most common chromosomal abnormality associated with HSCR is Down syndrome ,which occurs in 2%-10% of all individuals with HSCR.
Although individuals with Down syndrome are at a hundred-fold higher risk for HSCR than the general population , none of the established HSCR genes reside on chromosome 21; thus the association between trisomy 21 and HSCR remains unexplained.
(Hirschsprung disease: a genetic study,1985)
( Hirschsprung's disease: genetic and functional associations of Down's and Waardenburg syndromes,1998 )
Syndromes associated with HSCR are listed in alphabetical order; the prevalence of HSCR in each syndrome varies widely and is estimated in table:
( Hirschsprung Disease Overview,2011)
DIAGNOSIS
The diagnosis of HSCR requires histopathologic demonstration of absence of enteric ganglion cells in the distal rectum.
HD is diagnosed based on symptoms and test results.
A doctor will perform a physical exam and ask questions about your child’s bowel movements. HSCR is much less likely if parents can identify a time when their child’s bowel habits were normal.
If HSCR is suspected, the doctor will do one or more tests.
X rays
An x ray is a black-and-white picture of the inside of the body. To make the large intestine show up better, the doctor may fill it with barium liquid. Barium liquid is inserted into the large intestine through the anus.
If HSCR is the problem, the last segment of the large intestine will look narrower than normal. Just before this narrow segment, the intestine will look bulged. The bulging is caused by blocked stool stretching the intestine.
Manometry
During manometry, the doctor inflates a small balloon inside the rectum. Normally, the rectal muscles will relax. If the muscles don’t relax, HSCR may be the problem. This test is most often done in older children and adults.
Biopsy
Biopsy is the most accurate test for HSCR. The doctor removes a tiny piece of the large intestine and looks at it with a microscope. If nerve cells are missing, HSCR is the problem.
(What I need to know about Hirschsprung Disease,2013 )
TREATMENT
Treatment of Hirschsprung's disease consists of surgical removal (resection) of the abnormal section of the colon, followed by reanastomosis.
- Pull-through Procedure
HSCR is treated with surgery called a pull-through procedure. A surgeon removes the segment of the large intestine lacking nerve cells and connects the healthy segment to the anus. The pull-through procedure is usually done soon after diagnosis.

- Ostomy surgery
An ostomy allows stool to leave the body through an opening in the abdomen. Although most children with HSCR do not need an ostomy, a child who has been very sick from HSCR may need an ostomy to get better before the pull-through procedure.
For ostomy surgery, the surgeon first takes out the diseased segment of the large intestine. The end of the healthy intestine is moved to an opening in the abdomen where a stoma is created. A stoma is created by rolling the intestine’s end back on itself, like a shirt cuff, and stitching it to the abdominal wall. An ostomy pouch is attached to the stoma and worn outside the body to collect stool. The pouch will need to be emptied several times each day.
If the surgeon removes the entire large intestine and connects the small intestine to the stoma, the surgery is called an ileostomy. If the surgeon leaves part of the large intestine and connects it to the stoma, the surgery is called a colostomy.
Later, during the pull-through procedure, the surgeon removes the stoma and closes the abdomen with stitches.
(What I need to know about Hirschsprung Disease,2013 )