Giulia Negro
Valentina Paonessa
DEFINITION
Cerebral cavernous malformation (CCM; OMIM 116860) is a vascular malformation, mostly located in the central nervous system.
CCMs are mulberry-like formations, consisting of vascular sinusoids with an apparently stagnant venous blood flow. The sinusoids are lined by a thin layer of endothelium devoid of mature vessel elements and embedded in a dense, multilayered basal lamina often surrounded by reactive gliosis and hemosiderin rim.
Parenchyma is never found within these lesions and the tight junctions between the endothelial cells are poorly formed or absent.

EPIDEMIOLOGY
- CCMs affect approximately 0,5% of population.
- They can occur in sporadic forms (80% of cases, usually with single lesions) but also as familial forms with an autosomal dominant condition with incomplete penetrance and variable clinical expression (20% of cases, usually with multiple lesions).
- They are the second most common vascular lesion behind developmental venous anomalies and account for 10-15% of all vascular malformations. Only 20%-30% of affected individuals develops symptomatic disease.
- Affected children present a bimodal pattern with peaks at 0-2 years of age and 13-16 years of age.
- The average age of adult presentation is in the 4th or 5th decade of life.
- The proportion of familial cases has been estimated to be as high as 50% in Hispanic-American patients of Mexican descent.
- In regard to sex, the prevalence of CCMs appears equal among men and women; however, some studies have raised the question of an increased incidence of symptomatic lesions in women (3,2% vs 0,9%).
Update on the natural history of cavernous malformations and factors predicting aggressive clinical presentation, 2010
SYMPTOMS
Cerebral cavernous malformations have a wide range of presenting symptoms, but the majority of lesions remains asymptomatic throughout life and is found incidentally.
| Localization | Symptoms |
| Supratentorial lesions | usually present with recurrent headaches and seizures. These seizures are thought to be induced by recurrent microhemorrhages from the CCM, resulting in perilesional gliosis and inflammation, both of which are epileptogenic |
| Infratentorial lesions | present with focal neurological deficits (for example cranial nerve palsy, hemiparesis and hemisensory deficits) or gait ataxia |
| Brainstem CCMs | may even associate with cardiovascular instability, respiratory compromise and hiccups |
All of the above symptoms can be found with or without acute hemorrhage. The annual bleeding rate ranges between 0,25% and 6%.
Update on the natural history of cavernous malformations and factors predicting aggressive clinical presentation, 2010
DIAGNOSIS
Clinical Diagnosis
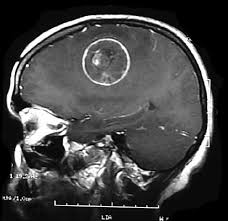
Usually, CCM lesions are almost angiographically silent and can be missed by computed tomography scanning. However, magnetic resonance imaging (MRI) is a particularly sensitive imaging tool for these lesions.
In most cases of MRI, they present with a peripheral T2 hypointensity due to hemosiderin deposition in the surrounding parenchyma and T2 GRE has been recognized as the gold standard for detection of CCMs.
MRI scans may be repeated over the course of the treatment to detect any changes in the size of malformation, recent bleeding and the appearance of new lesions.
With MRI imaging these lesions can be categorized into four types, according to the presence and pattern or absence of blood signal.
| Type | MR Signal | Histopathology | Clinical Correlation |
| Type-1 | −SE T1: hyperintense core
−SE T2: hyperintense core or hypointense core | Subacute hemorrhage | Acute hemorrhage; high frequency of bleeding relapse |
| Type-2 | −SE T1: reticulated mixed signal core
−SE T2: reticulated mixed signal core with surrounding hypointense rim | Lesions with hemorrhages and thromboses of varying ages | / |
| Type-3 | −SE T1: iso- or hypointensity
−SE T2: hypointense lesion with hypointense rim magnifying the size of the lesion | Chronic hemorrhage with hemosiderin staining within and around the lesion | / |
| Type-4 | −SE T1: not seen
−SE T2: not seen
−GRE: punctate hypointense lesion SWI: punctuate hypointense lesion | Tiny CCM or telangiectasia | Possibly represent true new lesions |
Specific MRI sequences and programs:
SE = spin echo MRI
GRE = gradient echo MRI
SWI = susceptibility-weighted imaging MRI
Genetic and cellular basis of cerebral cavernous malformations implications for clinical management, 2012
Molecular Genetic Testing
The diagnosis of familiar cerebral cavernous malformation (FCCM) can be confirmed by molecular genetic testing of the following three genes in which mutations are known to cause fCCM: KRIT1 (locus name CCM1), MGC4607 (locus CCM2) and PDCD10 (locus CCM3).
In general, patients requiring prenatal diagnosis have family members affected by severe symptoms.
One analytical approach to search for a pathogenetic mutation in the CCM family consists of the consequential sequence analysis of the coding regions as well as the exonic–intronic junctions in the three genes.
In the case of negative results, scientists suggest to proceed with multiplex ligation-dependent probe amplification (MLPA) analysis. MLPA, a variation of polymerase chain reaction (PCR) allows simultaneous quantification of several nucleic acid sequences in a single reaction. Amplification products are separated by sequence gel electrophoresis, and MLPA probes are able to distinguish between sequences that differ in only one base pair. This technique allows the identification of other kind of mutations such as genomic rearrangements (insertions or deletions).
MLPA technique has rapidly gained acceptance in genetic diagnostic laboratories for its simplicity compared with other methods, its relatively low cost, its high throughput, and its perceived robustness.
In the case of negative results from both approaches, another opportunity successfully applied is offered by using a combination of microsatellite marker analysis and real-time quantitative polymerase chain reaction (RT-qPCR).
While the former is based on a repeated sequence of DNA (often consisting of two, three or four nucleotides that can be repeated 10–100 times) used frequently in human genomics, RT-qPCR is the most sensitive technique for mRNA quantification currently available.
The presence of a causative mutation in one of the known genes is always reconfirmed by new sample followed by a new molecular procedure. On the contrary, if the disease-causing mutation cannot be detected there are two possible explanations: germline mosaicism in a parent or a de novo mutation in the proband.
Cerebral Cavernous Malformation, Familial, 2011
PATHOGENESIS
Familiar Cerebral Cavernous Malformation is caused by loss-of-function mutations in one of these three genes involved in blood vessels formation: CCM1 (also known as KRIT1), CCM2 (MGC4607, OSM, Malcavernin) and CCM3 (PDCD10, TFAR15, mapped to chromosome 3q25.2-q27 ).
CCM1, mapped to chromosome 7q21-22

CCM2, mapped to chromosome 7p15-p13

Mutations in the gene encoding KRIT1, a Krev-1/rap1a binding protein, cause cerebral cavernous malformations (CCM1), 1999.
Mutations in a gene encoding a novel protein containing a phosphotyrosinebinding domain cause type 2 cerebral cavernous malformations, 2003
Mutations in apoptosis-related gene, PDCD10, cause cerebral cavernous malformation 3, 2005
Furthermore, it is believed that a "second hit mutation" (Knudsen Hypothesis) is necessary for the onset of the disease: loss of one allele in all cells (germline mutation, first hit) would be followed in some cells by a somatic mutation in the other allele (second hit), but it is not clear if the second hit could affect genes other than CCM1, CCM2, CCM3, or epigenetic factors.

Biallelic somatic and germline mutations in cerebral cavernous malformations (CCMs): evidence for a two-hit mechanism of CCM pathogenesis, 2009
A two-hit mechanism causes cerebral cavernous malformations: complete inactivation of CCM1, CCM2 or CCM3 in affected endothelial cells, 2009
Two-hit mechanism in cerebral cavernous malformation? A case of monozygotic twins with a CCM1/KRIT1 germline mutation, 2013.
Patients with loss-of-function mutations in any of the three CCM genes (which are expressed in many cell types and tissues, not only in the brain) develop similar brain vascular lesions, nevertheless CCM3 might also act separately from KRIT1 and CCM2, as its mutation often results in a more severe form of the disease.
Patterns of expression of the three cerebral cavernous malformation (CCM) genes during embryonic and postnatal brain development, 2006
Genotype-phenotype correlations in cerebral cavernous malformations patients, 2006

The CCM proteins bind to each other in order to form a signalling platform around a ternary KRIT1–CCM2–CCM3 complex. CCM2 acts as the central hub, using two independent binding sites to simultaneously interact with KRIT1 and CCM3.
CCM1 and CCM2 protein interactions in cell signaling: implications for cerebral cavernous malformations pathogenesis, 2005.
CCM3 interacts with CCM2 indicating common pathogenesis for cerebral cavernous malformations, 2007
Novel CCM1, CCM2, and CCM3 mutations in patients with cerebral cavernous malformations: in-frame deletion in CCM2 prevents formation of a CCM1/CCM2/CCM3 protein complex, 2008
Enhanced permeability is a hallmark of CCM lesions, and much effort has been invested in understanding its molecular basis using in vitro and in vivo models of CCM1-, CCM2-, and CCM3-deficient endothelial cells. As a result, it is now clear that CCM proteins are important for the control of endothelial permeability through actin regulation and endothelial junction formation and maintenance.
In fact, these proteins interact also with proteins specifically involved in cell-cell adhesion, cell shape, polarity and cell adhesion to extracellular matrix.
Rap1 was the first reported Krit1 partner and it activates integrins and cell-cell adhesion by stimulating the formation and maintenance of adherens junctions. In cell-cell junctions Krit1 co-localizes with β-catenin and VE-cadherin for the maintenance of the endothelial barrier and this localization is dependent upon activation of Rap1. CCM1 depletion inhibits the association of VE-cadherin with β-catenin, and causes a concomitant increase in the presence and function of β-catenin in the nucleus, where it acts as transcription factor for genes involved in tumorigenesis.
Linking Rap to cell adhesion, 2005.
KRIT-1/CCM1 is a Rap1 effector that regulates endothelial cell cell junctions, 2007
Rap1 and its effector KRIT1/CCM1 regulate β-catenin signaling, 2010
In addition, KRIT1 binds ICAP1 (that plays a role in regulation of junctional stability by having an impact on ECM remodeling and integrin activation) and HEG1 (a transmembrane protein expressed almost exclusively by vascular endothelial cells that controls junctional stability), therefore a loss of CCM1 modifies the stabilization of endothelial junctions and increases the number of actin stress fibers, leading to cell shape changes.
CCM1-ICAP-1 complex controls beta1 integrin-dependent endothelial contractility and fibronectin remodeling, 2013
Structural basis of the junctional anchorage of the cerebral cavernous malformations complex, 2012
Changes in the conformation of KRIT1 are thought to regulate its localization: ICAP1 binding is associated with a presumed ‘open’ conformation, but it can also have a ‘closed’ conformation and this happens when CCM1 binds to microtubules. Unfortunately the functional and signaling importance of KRIT1 conformational changes has not yet been adequately explored.
Moreover, recent data identify the CCM complex as a scaffold for the Rho family GTPases RhoA, Rac and Cdc42, and for the MAPK (mitogen activated protein kinases) and STK (Ser/Thr kinases) kinases.
Given that the loss of CCM1, CCM2, or CCM3 protein results in similar pathological lesions, the common dysregulated biochemical pathway in endothelial cells is increased expression and activation of the small GTPase RhoA. The CCM complex seems to regulate RhoA activity by using the CCM2 PTB domain to recruit SMURF1, a RhoA ubiquitinase which acts as a key effector of the GTPases Cdc42 and Rac and the 1-PAR6-PKCζ polarity complex. In this way SMURF1 leads to RhoA degradation and regulates cell shape, polarity, cell-cell contact and motility. Studies using a well-characterized Förster Resonance Energy Transfer (FRET) biosensor for RhoA showed that the loss of CCM1, -2, or -3 results in an increase in RhoA activation in the endothelial cells. This activation leads to the induction of its downstream effector ROCK, a kinase which phosphorylates a series of proteins, the most important of which is myosin light chain, or MLC, a regulator of endothelial and SMC contractility. MLC phosphorylation increases contractility and results in cell–cell adhesion disruption and in vascular permeability alteration.
Rho and ROCK signaling in VEGF-induced microvascular endothelial hyperpermeability Microcirculation, 2006
Regulation of cell polarity and protrusion formation by targeting RhoA for degradation, 2003
Rho kinase inhibition rescues the endothelial cell cerebral cavernous malformation phenotype, 2010
CCM2 plays a scaffolding role between RAC1 and MEKK3 in the p38 MAP kinase cascade in response to osmotic stress. In fact, the activation of p38 MAPK regulates gene transcription and post-translational modification of cytoskeletal remodelling proteins when they are exposed to prolonged hyperosmotic stimulation, in order to maintain cellular integrity and homeostasis. Therefore, a loss of CCM2 could cause the disruption of cytoskeletal architecture. Nonetheless, further studies are still required to understand the role of the CCM complex in this pathway.
Rac-MEKK3-MKK3 scaffolding for p38 MAPK activation during hyperosmotic shock, 2003


The CCM2 PTB domain also binds TrkA, a receptor tyrosine kinase found on nerve cells. This interaction induces cell death in neuroblastoma or medulloblastoma, probably through the recruitment of a complex between CCM3 and STK25. How it induces cell death, and whether this pathway contributes to the CCM phenotype, remains unclear.
CCM2 mediates death signaling by the TrkA receptor tyrosine kinase, 2009
STK25 protein mediates TrkA and CCM2 protein-dependent death in pediatric tumor cells of neural origin, 2012
CCM3 directly interacts with the germinal center kinase III serine/threonine kinases STK24, STK25 and MST4 (MASK), three proteins involved in cell polarization and migration. The CCM3– GCKIII interactions are likely important for targeting STK24, STK25, and MST4 to specific locations in the cell, including the cell membrane, where they can phosphorylate the ERM proteins (ezrin, radixin, and moesin) that link the actin cytoskeleton to the membrane. STK24 and STK25 control endothelial cell-cell junctions by directly activating the moesin, a negative regulator of Rho; knockdown of moesin increases Rho activity in endothelial cells.
Structural mechanism of CCM3 heterodimerization with GCKIII kinases, 2013

The Ser/Thr kinases STK25 and MST4 were found to localize to the Golgi apparatus via an association with the Golgi resident protein GM130. Mislocalization of these kinases results in defects in Golgi positioning and cell migration. Recently, CCM3 was shown to participate in this effect by stabilizing the GCKIII proteins to promote Golgi orientation and assembly and proper cell orientation.
CCM3/PDCD10 stabilizes GCKIII proteins to promote Golgi assembly and cell orientation, 2010
Another plausible hypothesis to explain the contribution of CCM3 inactivation to the CCM pathogenesis is the involvement of CCM3–STK24 complex in preventing neutrophil degranulation: a loss of CCM3 or STK24 results in increased exocytosis, that may contribute to defective vessel tubular morphology observed in the CCM disease.
A network of interactions enables CCM3 and STK24 to coordinate UNC13Ddriven vesicle exocytosis in neutrophils, 2013
It seems also that abnormal intracellular levels of reactive oxygen species (ROS) promote extensive cellular damage and dysfunctions in endothelial cells, as well as in the pathogenesis of human vascular diseases, including cerebrovascular diseases: it represents an additive event involved in CCM pathogenesis. In particular, CCM1 regulates the expression of the enzyme SOD2 and of its transcription factor FoxO1 by protecting cells from oxidative stress; therefore, a loss of CCM1 leads to a significant increase in intracellular ROS levels and to oxydative stress.
KRIT1 regulates the homeostasis of intracellular reactive oxygen species, 2010
Very recent work has shown that another critical step in the formation of CCM lesion is the induction of the endothelial-to-mesenchymal transition (EMT), defined as the acquisition of mesenchymal- and stem-cell-like characteristics by the endothelium. The molecular basis for how this occurs is not yet understood, but it seems likely that the CCM complex plays an important role in direct regulation of SMAD proteins, whose activation is increased by loss of CCM1. Loss of CCM1 specifically mediates the upregulation of endogenous BMP6 which activates the transforming growth factor-β (TGF-β) and bone morphogenetic protein (BMP) signalling pathway, that induces EMT and, as a consequence, the development of vascular malformations, altered junction organization, loss of cell polarity, increased cell proliferation and migratory capacity.
CCM1 acts also as a Notch activator, that abrogates BMP6 expression. In this case loss of CCM1 mediates Notch inhibition, that upregulates BMP6 expression in the brain endothelium: BMP6 activity contributes to the observed EndMT.
EndMT contributes to the onset and progression of cerebral cavernous malformations, 2013
Furthermore, CCM1 induces HEY1 and HEY2, direct targets of NOTCH signaling, and the NOTCH ligand DLL4. Active NOTCH signaling inhibits endothelial proliferation, migration, sprouting and branching. CCM1 promotes AKT phosphorylation (that occurs after interaction with phosphatidylinositol trisphosphates (PIP3), a protein generated from PIP2 which in turn interacts with CCM proteins) in a NOTCH-dependent and independent manner. Moreover, it inhibits ERK1/2 phosphorylation indirectly through activation of the DELTA-NOTCH cascade. CCM1-loss-of-function causes the opposite effects with excessive sprouting angiogenesis and cellular proliferation.
Cerebral cavernous malformation protein CCM1 inhibits sprouting angiogenesis by activating DELTA-NOTCH signaling, 2010

A central question is why CCM lesions are mostly localized in the brain vasculature although the gene is inactivated in all types of endothelial cell. The brain vasculature requires a continuous crosstalk with astrocytes to maintain the organization of the neurovascular unit. The lack of an appropriate interaction with neural cells may strongly influence the development of lesions, since this interaction is unique for the brain vessels and may explain why in other organs the appearance of vascular malformations is much less frequent. It is a possibility, but further studies are necessary before we can link the known functions of CCM proteins to CCM formation.
Loss of cerebral cavernous malformation 3 (Ccm3) in neuroglia leads to CCM and vascular pathology, 2011
Aminoacids Percentage Of CCM Proteins

THERAPY
- Surgical technique: lesionectomy could refer both to the ‘pure’ resection of cavernoma (disregarding the hemosiderin ring and whatever adjacent cortical epileptiform abnormalities) or to the combined resection of all visible structural abnormalities, comprising the cavernoma and the hemosiderin ring.
- Possible additional drugs:
- Fasudil, a Rho kinase inhibitor, is able to reduce development and hemorrhagic rates of CCM lesions in a mouse model of CCM-1 disease compared with placebo controls. Lesions in treated animals were smaller and less likely associated with inflammation and endothelial proliferation and exhibited decreased expression of Rho kinase activation biomarkers.
- Sorafenib can ameliorate loss of CCM1-induced excessive microvascular growth, by reducing the microvessel density to levels of normal wild-type endothelial cells; in this way, it could be a potential therapeutic approach also for humans.

- Statin drugs may be used for treating CCM through the inhibition of Rho GTPases. There is evidence that the Rho GTPase pathway can be directly activated by ROS. Besides inhibition of Rho GTPases, the serum cholesterol-lowering drug statin exerts powerful intracellular antioxidant activities in endothelial cells, including the inhibition of superoxide production and the improvement of ROS scavenging.
- Antioxidant compounds may be safer than statins thanks to their low side effects and thus may be used for treating CCM disease.
- Analgesics and antiepileptics are employed for treating headache and epilepsy, respectively.
Genetic and cellular basis of cerebral cavernous malformations implications for clinical management, 2012
Evaluating strategies for the treatment of cerebral cavernous malformations, 2010
Fasudil decreases lesion burden in a murine model of cerebral cavernous malformation disease, 2012