Introduction
Hereditary multiple exostoses (HME or MHE), also known as multiple osteochondromatosis (MO) or diaphyseal aclasis is a genetic bone disorder in which cartilage outgrowths called exostoses or ostechondromas form next to the growth plate of skeletal elements (such as long bones, ribs and pelvis) and protrude into the adjacent perichondrium and neighboring tissues.
The normal growth plate, located at the epiphysis of long bones, has a highly organized structure. In contrast, the osteochondromas of HME consist of a disorganized collection of chondrocytes leading to random ossification.
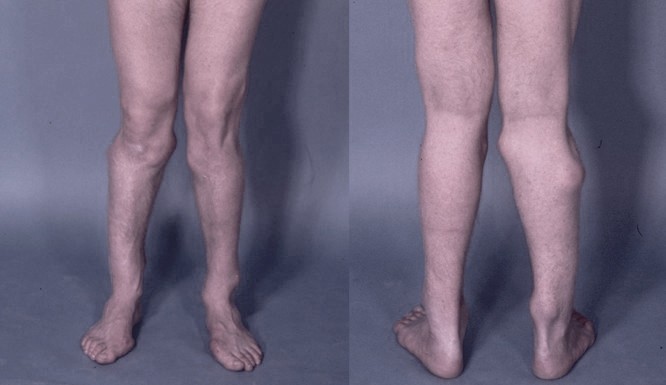
Photograph of the legs of a 26-year-old male showing multiple lumps leading to deformity
HME typically presents early during postnatal life with most affected individuals (>80%) being diagnosed by the age of 10. Osteochondromas develop and increase in size in the first decade of life, ceasing to grow when the growth plates close at puberty. No new lesions develop thereafter, although regrowth of an exostoses after surgical removal is possible.
Exostoses can vary widely in size and shape; they may be:
* pedunculated: broccoli-like appearance with a stalk and growth towards the end of stalk.
* sessile: broad-base attachment to the outer bone, called the cortex. No stalk is present.
The severity of this disease varies significantly within and between families: some patients may have as few as two tumors, but most patients develop many more and the numbers of tumors can run into the hundreds. The majority are asymptomatic and located predominantly in femur, tibia and fibula, but the humerus, radius, ulna, ribs, scapula and bones of the hands and feet are also often involved. Facial bones are not affected.

HME can lead to a variety of orthopaedic deformities, like deformities of the forearm (shortening of the ulna with secondary bowing of radius), inequality in limb lenght, deformity of the ankle and disproportionate short stature.
Depending on their location, the exostoses can cause the following problems: pain or numbness from compression of peripheral nerves (paraesthesiae), vascular compromise (a rupture of the popliteal artery has often been described), irritation of tendon muscle resulting in loss of motion, early onset arthritis, Madelung's syndrome as well as a limited range of motion at the joints upon which they encroach. HME patients may have abnormal scar formation. Moreover, osteochondromas bear the risk for fracture of the bony stalk during physical exercise. Exostoses may be visible to naked eye from outside and appear to slow their rate of growth when they reach a certain, variable mass.
The most important complication of HME is malignant transformation of an osteochondroma, which is estimated to occur in 0.5-5% of patients. Clinical signs of malignant transformation include an increase in size and pain. Malignant transformation of osteochondroma leads to a secondary peripheral chondrosarcoma in 94% of the cases. The suspicion of secondary chondrosarcoma is indicated by growth of the tumor after puberty, the presence of pain or a thickness over 1 cm of the cartilaginous cap in adults.
Multiple osteochondromas, 2008
Hereditary multiple exostoses, 1991
Manifestation of HME, 2005
Epidemiology
The prevalence of HME is estimated at 1:50000 people within the general population and seems to be higher in males (male to female ratio 1,5:1). This is probably do to the fact that females tend to have a milder phenotype and are therefore more easily overlooked. Approximately 62% of the patients with multiple osteochondromas have a positive family history.
HME has a 96% penetrance, which means that if the affected gene is indeed transmitted to a child, the child will have a 96% chance of actually manifestating the disease and 4% chance of having the disease but never manifesting it. Other studies have observed both incomplete and variable penetrance but without calculating the % penetrance.
Pathogenesis
HME is an autosomal dominant disorder: this means that a patient with HME has a 50% chance of transmitting that disorder to his/her children. Most individuals with HME have a parent who also has the condition, but approximately 10-20% of HME patients have the condition as a result of a spontaneous mutation and are thus the first person in their family to be affected.
Since HME is a dominant disorder, apparently loss of only one allele is sufficient to cause disease.
In almost 90% of HME patients germline mutations has been linked with two main genes:
* EXT1 which maps to chromosome 8q24.1
* EXT2 which maps to chromosome 11p13
Addiotional linkage to the short arm of chromosome 19 has been found, suggesting the existence of an EXT3-gene, although the gene has never been identified.
EXT1 and EXT2 are highly similar: the EXT1 gene was reported to show linkage in 44-46% of HME families, whereas EXT2 would be involved in 27%. Mutations in EXT1 seem associated with a more severe phenotype as compared to EXT2.

The majority of HME cases arises from missense or frameshift mutations in EXT1 and EXT2, leading to synthesis of abnormally truncated forms of the EXT proteins. Interestingly, while EXT1 mutations occur throughout the entire length of the gene, mutations in EXT2 concentrate towards its N-terminus, implying specific functions for this part of the protein.
EXT1 and EXT2 have been identified as tumor suppressor genes since loss of heterozygosity at these loci occurs in HME patients whose benign tumors transform into chondrosarcomas. These genes encode Golgi-associated glycosyltransferases (exostosin-1 and exostosin-2) that are responsible for the synthesis of heparan sulfate (HS), a key component of cell surface-associated HS-rich proteoglycans (HSPGs) such as syndecan and glypican and matrix-associated proteoglycans including perlecan. Both EXT1 and EXT2 are needed for HS synthesis since the proteins form multimeric complexes in the Golgi. The HS chains of HSPGs impact a number of signaling proteins critical to skeletal development, such as Indian hedgehog (IHh), Fibroblast Growth Factor (FGFs), Bone Morphogenetic Proteins (BMPs) and Wnt. The HSPGs can function as co-receptors for some of these proteins, but can also influence their distribution, range of action, stability and action on target cells. In general, the HME-associated phenotypes are widely believed to arise from impaired HS synthesis and accompanying HS deficiency in the skeleton and other tissues and organs. However, it remains unclear how the HS deficiency alters cell signaling within the cartilage and/or surrounding perichondrium in developing and growing skeletal elements, as well as, whether it may also affect other processes and events leading to the clinical HME phenotypes.
MHE: elucidating the pathogenesis through interdisciplinary research, 2014
Heparan sulfate polymerization, 2002
Heparan sulfate in skeletal development, growth and pathology, 2013
Diagnosis

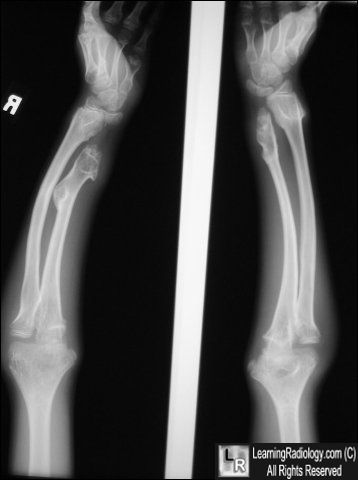
When a patient is suspected to have HME, the full radiological and clinical documentation, histology (if available), patient history and family history have to be carefully reviewed.
Given the specific radiological and histological expertise needed and the rarity of the disorder and of those in the differential diagnosis, it is recommended that this review is permofed by specialists in the field.
If this review is indicative for HME, the peripheral blood of the patient may be screened for germline in EXT1 or EXT2. With the currently used methods it is possible to detect point mutations or gross deletions in almost 90% of HME patents. If the exact mutation is known, antenatal diagnosis is technically possible for individuals with HME who are considering starting a family.
Technetium skeletal scintigrams are occasionally used to determine number of exostoses.
In case of a positive family history in which HME is clearly established in relatives, the diagnosis in HME can be clinically made and mutation analysis is not essential.
To evaluate possible malignant transformation in case of complaints or growth of the lesion after puberty, the size of the cartilaginous cap can be weel established with T2-weighted magnetic resonance imaging. A cartilage cap >1.5 cm should be regarded with caution.
Diagnosis
Radiographic features
Trevor's disease, metachondromatosis and Ollier disease are considered in the differential diagnosis of solitary and hereditary osteochondromatosis.
Treatment
If the diagnosis of Hereditary Multiple Exostoses is established, patients should be well instructed to seek earlier medical attention if their condition changes, for example if there is pain or growth of a known lesion. It is important to realize that no new osteochondromas develop after puberty. Moreover, regular follow-up to discover potential malignant transformation at an early stage to enable adequate treatment should be considered.
After base-line documentation, one should consider screening patients regularly, although there are yet no studies available that have proven efficacy of screening. Exostoses are benign and do not affect life expectancy. Surgical excision is performed only when exostoses lead to growth disturbances and function complaints or lead to disability. Knee osteotomies are associated with high incidence of peroneal nerve paralysis.
Surgery, physical therapy and pain management are currently the onyl options available to HME patients, but success varies from patient to patient and many struggle with pain, fatigue and mobility problems throughout their lives. It is not uncommon for HME patients to undergo numerous surgical procedures (sometimes greater than 20) to remove painful or deforming exostoses, correct limb length discrepancies or improve range of motion, most during childhood and adolescence. Bigger challenges arise from the forearms and lower legs (below the knee), where the impact on linear growth of bones can differentially affect one bone more than the other, leading to tethering, bowing and angular growth deformities. While surgical interventions have been described for these scenarios, each is fraught with complications.
If lesions change over time, further examination, using magnetic resonance (MR) imaging including contrast enhanced MR sequences is indicated.
In case of malignancy, en-bloc resection of the lesion and its pseudocapsule with tumor-free margins, preferably in a bone tumor referral center, should be permormed, resulting in excellent long term clinical and local results. The most common location is however the pelvis where the large cartilage cap can be difficult to excise.
Treatment
Possible connection to autism
Some parents of children with HME have observed autism-like social problems in their children. To explore those observations more deeply, a 2012 study used a mouse model of HME to observe cognitive function. The findings indicated that the mutant mice endorsed three autistic characteristics: social impairment, language deficits and repetitive behaviour.
Autism-like socio-communicative deficits and stereotypies in mice lacking heparan sulfate, 2012
Other references
Hereditary multiple exostoses
Diaphyseal aclasis
Multiple exostoses, type I