Non è così facile misurare il glicogeno nei tessuti
Ultrastructural changes during in situ early postmortem autolysis in kidney, pancreas, liver, heart and skeletal muscle... 2004
High Glycogen Levels in Brains of Rats With Minimal
Environmental Stimuli: Implications for Metabolic Contributions
of Working Astrocytes 2002
Glycogen synthesis
Glycogen Synthase (GS)
GS degradation

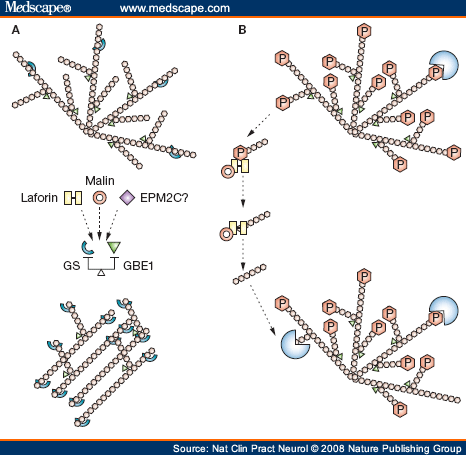
Two recent theories of the pathogenesis of Lafora bodies. (A) GS (light blue) extends the growing glycogen chain in a linear manner. GBE1 (green) branches the linear glycogen, giving the molecule its spherical shape. If the balance between extension and branching is disrupted, poorly branched glycogen, or polyglucosans, form instead of normal glycogen. Laforin, malin, and, possibly, other proteins involved in Lafora disease regulate the balance of the two enzymes, preventing polyglucosan formation. (B) Glycogen is normally phosphorylated, and this hinders its degradation. Laforin (yellow) dephosphorylates glycogen. Laforin must then be removed to enable degradation to start, and this is accomplished by malin (red circle). If laforin or malin is not present, glycogen cannot be degraded and, over time, it loses its spherical structure, accumulating as polyglucosans to form Lafora bodies. Abbreviations: GBE1, glycogen branching enzyme; GS, glycogen synthase; P, phosphate.

(a) Under normal circumstances, astrocytes synthesize and store glycogen. Neurotransmitters such as noradrenaline (NA), vasoactive intestinal peptide (VIP), adenosine (ADE) or ATP trigger the breakdown of glycogen to enable the release of lactate, which is taken up as fuel by neurons. In neurons, glycogen synthase (GS) is hyperphosphorylated and thereby inactivated. The malin-laforin complex targets hyperphosphorylated GS for ubiquitin proteasome–dependent degradation. No glycogen is synthesized in neurons. (b) In Lafora disease, either malin or laforin is mutated. Thus, hyperphosphorylated GS escapes degradation and can be activated by dephosphorylation. Vilchez et al.7 show that either inhibition of glycogen synthase kinase by LiCl or by overexpression of the protein phosphatase 1 regulatory subunit PTG can activate GS in neurons, leading to the accumulation of abnormally branched glycogen species that are toxic to neurons (as indicated by the dashed cell outline.)
Glycogen: a Trojan horse for neurons 2007
Neural activity leads to the mobilization of energy from glycogen in astrocytes. A new paper reports that neurons have an ambivalent relationship with glycogen: they can synthesize it themselves, but that synthesis induces apoptosis. Presumably for this reason, neurons normally inhibit glycogen synthesis through two redundant pathways.
the malin-laforin complex markedly decreases PTG and glycogen synthase protein levels, thus inactivating the glycogen-synthesizing machinery, through a mechanism mediated by activation of the ubiquitin-proteasome pathway.
- The NHLRC1 gene encodes malin, a single subunit E3 ubiquitin (UBB; MIM 191339) ligase, which contains a RING-HC-type zinc finger and 6 NHL domains and is subclassified as a member of the RING-HCa family (Gentry et al., 2005 (PubMed 15930137)).(supplied by OMIM)


Progressive myoclonus epilepsy of the Lafora type (Lafora disease, LD, OMIM 274780) is a rare disease characterized by the presence of neurodegeneration, epilepsy and accumulation of poorly branched polyglycosans in different tissues. This disease leads to the death of the patient around 10 years from the onset. At the moment only a limited knowledge about the molecular bases of the disease is known and there is no available treatment. LD is a recessive autosomic disease and so far two genes have been described to be involved in the pathogenesis of the disease: EPM2A and EPM2B. EPM2A codes for a protein called laforin which has a carbohydrate binding domain (CBD) at the N-terminus and a dual specificity phosphatase (DSP) domain at the C-terminus. EPM2B codes for a protein called malin (with E3-ubiquitin ligase activity), which has a RING finger domain at the N-terminus and six NHL domains at the C-terminus involved in protein-protein interaction. Recently, our group has described that laforin and malin form a functional complex, suggesting that both proteins participate in a common physiological process and justifying why patients with mutation in either EPM2A or EPM2B are neurologically and histologically indistinguishable. One of the functions of this complex is to downregulate the levels of R5/PTG, a protein involved in the upregulation of glycogen synthesis. This function would explain why in the absence of a functional laforin-malin complex, cells would accumulate polyglycosans (a pathological determinant of the disease). In the complex, laforin acts as a targeting subunit of malin, being able to recognize different substrates that eventually will be ubiquitinated by malin and targeted for degradation. In addition to the role that laforin and malin may have on the regulation of glycogen synthesis, these proteins have alternative roles in cellular physiology. For this reason, we are studying the involvement laforin and malin in the regulation of different signalling pathways, such as those involving AMPK. These new regulatory functions of laforin and malin will help us to understand the pathophysiology of the disease and to define possible therapeutic targets.
Hum Mol Genet. 2009 Feb 15;18(4):688-700. Epub 2008 Nov 25.
The malin-laforin complex suppresses the cellular toxicity of misfolded proteins by promoting their degradation through the ubiquitin-proteasome system.
Garyali P, Siwach P, Singh PK, Puri R, Mittal S, Sengupta S, Parihar R, Ganesh S.
Department of Biological Sciences and Bioengineering, Indian Institute of Technology, Kanpur, India.
Lafora disease (LD), a progressive form of inherited epilepsy, is associated with widespread neurodegeneration and the formation of polyglucosan bodies in the neurons. Laforin, a protein phosphatase, and malin, an E3 ubiquitin ligase, are two of the proteins that are defective in LD. We have shown recently that laforin and malin (referred together as LD proteins) are recruited to aggresome upon proteasomal blockade, possibly to clear misfolded proteins through the ubiquitin-proteasome system (UPS). Here we test this possibility using a variety of cytotoxic misfolded proteins, including the expanded polyglutamine protein, as potential substrates. Laforin and malin, together with Hsp70 as a functional complex, suppress the cellular toxicity of misfolded proteins, and all the three members of this complex are required for this function. Laforin and malin interact with misfolded proteins and promote their degradation through the UPS. LD proteins are recruited to the polyglutamine aggregates and reduce the frequency of aggregate-positive cells. Taken together, our results suggest that the malin-laforin complex is a novel player in the neuronal response to misfolded proteins and could be potential therapeutic targets for neurodegenerative disorders associated with cytotoxic proteins.
J Biol Chem. 2008 Dec 5;283(49):33816-25. Epub 2008 Oct 13.
Abnormal metabolism of glycogen phosphate as a cause for Lafora disease.
Tagliabracci VS, Girard JM, Segvich D, Meyer C, Turnbull J, Zhao X, Minassian BA, Depaoli-Roach AA, Roach PJ.
Department of Biochemistry and Molecular Biology, Indiana University School of Medicine, Indianapolis, Indiana 46202, USA.
Lafora disease is a progressive myoclonus epilepsy with onset in the teenage years followed by neurodegeneration and death within 10 years. A characteristic is the widespread formation of poorly branched, insoluble glycogen-like polymers (polyglucosan) known as Lafora bodies, which accumulate in neurons, muscle, liver, and other tissues. Approximately half of the cases of Lafora disease result from mutations in the EPM2A gene, which encodes laforin, a member of the dual specificity protein phosphatase family that is able to release the small amount of covalent phosphate normally present in glycogen. In studies of Epm2a(-/-) mice that lack laforin, we observed a progressive change in the properties and structure of glycogen that paralleled the formation of Lafora bodies. At three months, glycogen metabolism remained essentially normal, even though the phosphorylation of glycogen has increased 4-fold and causes altered physical properties of the polysaccharide. By 9 months, the glycogen has overaccumulated by 3-fold, has become somewhat more phosphorylated, but, more notably, is now poorly branched, is insoluble in water, and has acquired an abnormal morphology visible by electron microscopy. These glycogen molecules have a tendency to aggregate and can be recovered in the pellet after low speed centrifugation of tissue extracts. The aggregation requires the phosphorylation of glycogen. The aggregrated glycogen sequesters glycogen synthase but not other glycogen metabolizing enzymes. We propose that laforin functions to suppress excessive glycogen phosphorylation and is an essential component of the metabolism of normally structured glycogen.
 Papers laforin
Papers laforin
Mol Cell Biol. 2008 Dec;28(23):7236-44. Epub 2008 Sep 29.
Laforin negatively regulates cell cycle progression through glycogen synthase kinase 3beta-dependent mechanisms.
Liu R, Wang L, Chen C, Liu Y, Zhou P, Wang Y, Wang X, Turnbull J, Minassian BA, Liu Y, Zheng P.
Department of Surgery, Division of Immunotherapy, Program of Molecular Mechanism of Diseases andComprehensive Cancer Center, University of Michigan, Ann Arbor, Michigan 48109, USA.
Glycogen synthase kinase 3beta (GSK-3beta) represses cell cycle progression by directly phosphorylating cyclin D1 and indirectly regulating cyclin D1 transcription by inhibiting Wnt signaling. Recently, we reported that the Epm2a-encoded laforin is a GSK-3beta phosphatase and a tumor suppressor. The cellular mechanism for its tumor suppression remains unknown. Using ex vivo thymocytes and primary embryonic fibroblasts from Epm2a(-/-) mice, we show here a general function of laforin in the cell cycle regulation and repression of cyclin D1 expression. Moreover, targeted mutation of Epm2a increased the phosphorylation of Ser9 on GSK-3beta while having no effect on the phosphorylation of Ser21 on GSK-3alpha. In the GSK-3beta(+/+) but not the GSK-3beta(-/-) cells, Epm2a small interfering RNA significantly enhanced cell growth. Consistent with an increased level of cyclin D1, the phosphorylation of retinoblastoma protein (Rb) and the levels of Rb-E2F-regulated genes cyclin A, cyclin E, MCM3, and PCNA are also elevated. Inhibitors of GSK-3beta selectively increased the cell growth of Epm2a(+/+) but not of Epm2a(-/-) cells. Taken together, our data demonstrate that laforin is a selective phosphatase for GSK-3beta and regulates cell cycle progression by GSK-3beta-dependent mechanisms. These data provide a cellular basis for the tumor suppression activity of laforin.