Francesca Bar & Tiziana Ruggiero
Nitric oxide and apoptosis
NO
Endogenous nitric oxide (NO) is synthesized from L-arginine by a family of NO synthase (NOS) isoenzymes: endothelial NOS (eNOS), neuronal NOS (nNOS) and inducible NOS (iNOS).
Generally, constitutive NOS (cNOS), such as eNOS and nNOS, is activated by a transient increase in cytosolic calcium which promotes the release of NO over several minutes. NOS is expressed in many cells, including macrophages and hepatocytes, after stimulation of immunological or inflammatory reactions, producing large amounts of NO for up to several days.
Alternatively cells can dispose of NO provided by NO-releasing compounds (NO donors) such as organic nitrates and S-nitrosothiols are valuable tools.
NO is a diffusible multifunctional transcellular messenger that has been implicated in numerous physiological and pathological conditions.
The biological activities of NO can be divided into:
• cGMP-dependent
• cGMP-independent pathways.
NO is a transducer of the vasodilator message from the endothelium to the vascular smooth muscle. It is also a neurotransmitter in the central and peripheral nervous systems, and participates in non-specific immune responses.
Although NO can affect the cellular functions through post-translational modifications of proteins both directly (nitrosylation and nitration) and indirectly (methylation and ribosylation), the main physiological signaling pathway of NO is considered to be the activation of guanylate cyclase, with consequential formation of cGMP and concomitant protein phosphorylation.
Afterwards cellular responses arise through cGMP-dependent signaling pathways, such as protein kinases (PKG), phosphatases and phosphodiesterases. However, there is growing evidence that the effects of NO on cell signaling pathways are not restricted to cGMP-dependent processes. NO has been shown to either promote or prevent apoptosis in numerous cell culture models in response to a diverse array of intracellular and extra-cellular proapoptotic stimuli. The mechanisms involve a modulation of either mitochondrial or non-mitochondrial apoptotic pathways either dependently or independently of cGMP or p53.
Apoptosis
Apoptosis, or programmed cell death, is the orchestrated collapse of a cell, staging membrane blebbing, cell shrinkage, protein fragmentation, chromatin condensation and DNA degradation followed by rapid engulfment of corpses by neighbouring cells. Apoptosis is biologically initiated by ligation of specific receptors of the tumor necrosis factor (TNF-R) family, such as CD95/Fas/Apo-1, TNFR1, and the receptor for TRAIL.
Ligand binding of the trimerized receptor recruits intracellular adaptor molecules like FADD and TRADD to form death-inducing signaling complex (DISC) and activates caspase-8, which in its turn starts the downstream of apoptotic signal cascade.
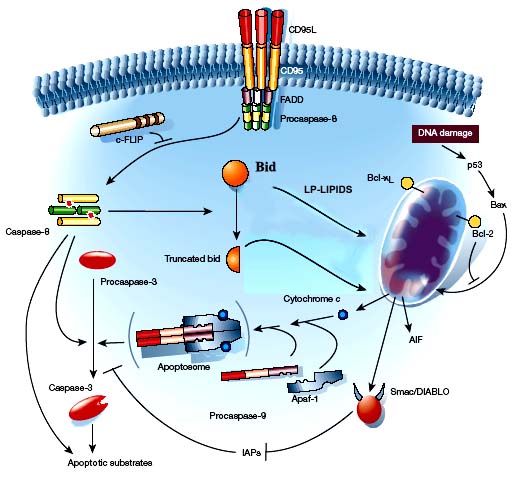
Mitochondria play a central role in apoptosis when the pro-apoptotic stimuli represent the so-called ‘intrinsic factors’, including oxidative stress, trophic factor withdrawal, DNA damage, and heat shock among the others.
Mitochondrial-mediated apoptotic signaling involves the release of several apoptogenic proteins from the intermembrane space of these organelles. Examples of such apoptosis-inducing factors (AIFs) are cytochrome c, smac/DIABLO and flavoprotein AIF. Cytochrome c-dependent apoptosis involves the recruitment of procaspase-9, apaf-1 and dATP into the mitochondrial apoptosome. This assembly is capable of activating caspase-3 with downstream activation of the caspase cascade.
Although the cell type specificity, the nature of the pro-apoptotic stimuli and the concentration of NO are very important factors, it is not correct to attribute the ambivalent behaviour of NO on apoptosis only to these variables, while the very common denominator is the cellular environment, with its composition and dynamic nature.
NO: proapoptotic inducer or antiapoptotic modulator
One immunological function of NO has been revealed to be the induction of cytotoxicity against tumor cells and surrounding tissues.
High concentrations of NO or peroxynitrite induce apoptotic cell death, in several cell types such as macrophages, thymocytes, pancreatic islets, certain neurons and tumor cells. Most of the proapoptotic effects of NO on these cells seem to be independent of cGMP. The factors affecting cell-specific sensitivity to NO-mediated apoptosis can be associated with the redox state and transition metal complexes within cells, and expression of survival genes.
Therefore, the threshold of NO level that triggers apoptosis is different from one cell to another.
Although NO promotes apoptosis in certain cells, it is apparent that NO displays antiapoptotic properties in other cell types, like hepatocytes. NO can protect these typed cells from apoptosis induced by many different types of stimuli such as:
• TNFa
• oxidative stress
• serum or glucose deprivation.
The biochemical mechanism underlying NO-mediated antiapoptotic effects may depend on cell type specifics with multiple signal pathways.
Proapoptotic pathways
• ACTIVATION OF MITHOCONDRIAL APOPTOTIC PATHWAY
NO impairs electron flux through the respiratory chain with the inhibition of multiple sites.
• At low physiological concentrations (10 nM – 1 μM), NO inhibits:
1. cytochrome oxidase (Complex IV) , through oxidation of the heme group (O2 binding domain) of cytochrome aa3 and possibly CuB+ . The reversible inhibition is competitive with O2, showing a dependence on the sub-cellular O2 tension.
2. the ubiquinone-bc1 segment (Complex III) of the respiratory chain, with consequent autooxidation of ubisemiquinone and subsequent generation of O2•– and thus H2O2.
• At higher concentrations (>1 μM), NO can directly oxidize ubiquinol, promoting ubisemiquinone auto-oxidation.
The fate of O2•– generated by these reactions shows a concentration-dependent regulation by NO:
• at a low [NO]ss superoxide dismutation to H2O2 is favored
• at a high [NO]ss the conversion to ONOO– is favored (due to the diffusioncontrolled reaction between NO and O2•– . Peroxynitrite itself can oxidize ubiquinol, amplifying O2•– generation and, potentially, its own formation.
The NO-induced generation of O2•– and ONOO– leads to the selective and persistent inhibition of NADH: ubiquinone reductase (Complex I) activity, but not Complex II and III.
Peroxynitrite also:
• causes irreversible damage to mitochondrial ATP synthase (Complex V) and aconitase
• promotes the permeability transition, cytochrome c release and apoptosis.
At a first approach, the pro- versus anti-apoptotic effect of NO may be related to the aforementioned sensitivity and dependence of oxyradical formation, mitochondrial respiration and ATP generation to the mitochondrial [NO]ss. At low concentrations, NO causes a transient, reversible de-energization of the mitochondria by affecting respiration, but not ATP generation. Prolonged exposure to NO or ONOO– can lead to the inhibition of ATP synthesis. A transient drop in ATP levels appeares to correlate with apoptosis, an energy-dependent process, whereas a persistent or complete decline in ATP resultes in necrosis.
• ACTIVATION OF CASPASE SIGNALING PATHWAY BY NO-INDUCED p53 EXPRESSION
Cytotoxic effects of NO and peroxynitrite on tumor cells have been proved to be the result of DNA damage which induces the accumulation of p53.
No-mediated p53 accumulation induces cell cycle arrest by p21 upregulation or apoptosis by increase in the ratio of Bax/Bcl-xL, cytochrome c release, and caspase activation.
• ACTIVATION OF JNK/SAPK AND p38 KINASE. MAP KINASE SIGNALING PATHWAYS
MAPKs transduce extracellular and intracellular signals into cellular responses by modulating gene and protein expression involved in cell differentiation, proliferation, survival and death.
Mammalian MAPKs can be divided into at least 3 groups, each containing isoenzymes:
• extracellular signal-related kinases (ERK1/2)
• c-Jun N-terminal kinases (JNK1/2/3)
• p38 kinases (p38α/β/γ/δ)
Mitogen-activated protein kinases (MAPKs) are integral components of cellular phosphorylation/de-phosphorylation signaling cascades. DNA damage, metabolic inhibitors, proinflammatory cytokines and growth factors, and a number of environmental stressors activate JNK and p38.
Typically ERK1/2 are associated with cell survival, whereas the SAPKs predominantly mediate apoptotic events. This has led to the term stress activated protein kinases (SAPKs).
MAPK are sensitive to oxidative and nitrosative stress arising from oxygen- and nitrogen-centered radicals, respectively. In turn, the cellular redox state and other components of the local environment can influence the sensitivity of MAPK to these reactive species. The sensitivity to NOx and H2O2 is enhanced by GSH depletion.
MAPK pathways are activated in a c-Ras-dependent or -independent manner.
1. The monomeric GTPbinding protein c-Ras (p21Ras) is an intermediate effector molecule in the transduction of signals from membrane receptor tyrosine kinases to the stimulation of MAP kinases, specifically ERK1/2. c-Ras appears to be a common signaling target of reactive free radicals, including NO and H2O2, and agents that modulate the cellular redox status such as GSH.
Importantly, the NMDA-mediated activation of ERK1/2 represents one of the rare cases in which this MAPK exerts pro-apoptotic effects.
2. Besides a direct effect on c-Ras, NO can also effect MAPK activation via a cGMP/PKG-dependent mechanism. Examples include the NO-mediated activation of the Ras-Raf1-MEK1-ERK1/2 pathway and downstream targets in HUVEC cells.
Rather than being limited to the exclusive activation of a specific MAPK, the initiation of apoptosis may result from a shift in the dynamic balance of activated ERK1/2 to activated JNK/p38. The cellular environment is likely to be an important determinant in the relative degree of activation of each MAPK.
• APOPTOSIS BY NO-MEDIATED CERAMIDE GENERATION
Sphingolipid metabolites, including ceramide, have been implicated as potential regulatory molecules in signal transductions that involve apoptotic cell death.
Apoptosis inducing stresses such as tumor necrosis factor-a, anti-Fas antibody, ionizing radiation, serum deprivation, anti-cancer drugs, heat shock, and hydrogen peroxide have been reported to increase intracellular ceramide. NO-induced mechanisms for the NO- apoptosis requires the generation of ceramide. NO donors increase cellular ceramide level, through the enhancement of magnesium-dependent neutral sphingomyelinase (N-SMase) activity and caspase-3 activity.
Ceramide formation can induce several apoptotic signal pathways, including:
• the release of mitochondrial cytochrome c into cytosol
• the activation of caspases-9 and -3
• JNK/SAPK activation
• the inhibition of protein kinase B/Akt
• the suppression of Bcl-2 expression.
Antiapoptotic pathways
• INHIBITION OF APOPTOTIC SIGNALING BY NO/cGMP PATHWAY
The NO-mediated activation of soluble guanylate cyclase produces the intracellular elevation of cGMP, which in turn activates PKG and at the same time decreases the cellular Ca2+ concentration, one of the key signals of apoptosis.
The interference of the NO/cGMP pathway with the apoptotic signal transduction is controversial.
Undoubtedly, cGMP production by NO can prevent apoptosis in some cell types including hepatocytes, neuronal PC12 cells, embryonic motor neurons, B lymphocytes, eosinophils, and ovarian follicles.
The antiapoptotic mechanisms of NO can be divided into cGMP-dependent and cGMP-independent mechanisms, which are likely cell-type specific.
In hepatocytes, PC12 cells and U937 cells with the antiapototic effects of NO are associated with cGMP production, which suppresses:
• the mitochondrial cytochrome c release
• ceramide generation,
• caspase activation.
• INHIBITION OF CASPASE ACTIVITY BY S-NITROSYLATION
Caspases (cysteine aspartic acid-specific proteases) are prominent among the death proteases and play critical roles in the initiation and execution of apoptosis. Typically, caspases are present in the cell as dormant zymogens.
Upon activation, caspases cleave a specific subset of cytosolic and nuclear targets resulting in protein degradation and DNA fragmentation. Pro-apoptotic stimuli activate caspases through several pathways, which may be inter-dependent.
Ultimately, initiation of the caspase enzyme cascade culminates in the activation of caspase-3, the execution enzyme of apoptosis.
An important type of mitochondrial-mediated apoptosis is the direct cytochrome c-dependent activation of caspase-3.
1. All caspases contain a single cysteine at the enzyme catalytic site. This thiol is susceptible to redox modification and can be effectively modified by S-nitrosylation in the presence of NO.
The ability of NO donors to prevent apoptosis in HUVEC cells over expressing caspase-3 was associated with a direct S-nitrosation of caspase-3 at the active site cysteine-163. Other studies found that in human lymphocyte cell lines the inactive caspase-3 zymogen is also S-nitrosated at the same critical cysteine residue under conditions of a basal steady-state production of NO by endogenous NOS activity.
Fas-induced apoptosis in these cells involved the promotion of caspase activation by a dual process:
• de-nitrosation of the cysteine residue
• cleavage of the zymogen to the active protease.
The active caspase-3, in turn, could be further inhibited by S-nitrosation following an increase in the steady-state level of NO.
2. The cleavage of Bcl-2 and cytochrome c release occurred downstream of caspase-3 activation, which typically requires the formation of the mitochondrial apoptosome after cytochrome c release.
The anti-apoptotic proteins Bcl-2 and Bcl-xL are substrates of caspases, with proteolytic cleavage generating pro-apoptotic ‘Bax-like fragments’. Bax and ‘BH-3 death domaincontaining’ members of the Bcl-2 family (e.g. Bak and Bid) are believed to promote cytochrome c release through formation of pores in the outer mitochondrial membrane.
Studies have demonstrated that NO inhibits cleavage of Bcl-2 by caspases in vitro through S-nitrosation.
In summary, NO can inhibit ‘initiator’ caspases, ‘executor’ caspases and the positive feedback amplification of apoptotic signaling arising from the downstream promotion of cytochrome c release by pre-activated caspase-3.
• REGULATION OF ANTIAPOPTOSIS-RELATED GENES EXPRESSION BY NO
NO and reactive nitrogen intermediates can interact with many different types of biomolecules including glutathione, iron-containing proteins, and tyrosine residue of protein, thereby changing the cellular redox potential and some signaling events. These oxidative and nitrosative stresses result in several gene expressions that modulate apoptosis.
We observed that NO potentially induces cytoprotective proteins such as HSP70 and HSP32 (heme oxygenase), which protect hepatocyte from apoptosis induced by TNFa, and oxidative or nitrosative stress.
MAPK SIGNALING AND MITHOCONDRIAL-DEPENDENT APOPTOSIS
MAPK signaling can modulate mitochondrial-mediated apoptotic pathways, predominantly through JNK.
JNK activation is not required for death-receptor-mediated apoptosis, but is required for mitochondrial cytochrome c release and the downstream activation of caspase-9 and -3 in response to diverse pro-apoptotic stimuli, including oxidative stress.
The molecular mechanism is unclear, but may involve regulation of the expression and phosphorylation state of the Bcl-2 family proteins assembled in the mitochondrial outer membrane.
For example, activation of JNK by diverse apoptotic stimuli is associated with the down-regulation of the anti-apoptotic Bcl-2 and Bcl-xL proteins and upregulation of the pro-apoptotic Bax and Bad proteins. Bcl-2 and Bcl-xL are substrates for JNK, with phosphorylation leading to inactivation. JNK-catalyzed phosphorylation and stabilization of p53 allows this proapoptotic protein to impair mitochondria function and induce the permeability transition through its mediator, p53-regulated apoptosis-inducing protein 1 (p53AIP1), a mitochondrial protein.
In contrast, the ERK1/2 pathway can prevent mitochondrial- mediated apoptotic events through the phosphorylation and activation of mitogen and stress activated kinase 1 (MSK1) and pp90 ribosomal S6 kinase (RSK).
RSK can phosphorylate and inactivate BAD, and both RSK and MSK1 are potent activators of the cAMP element binding protein (CREB), a transcription factor for Bcl-2.
References
Nitric oxide as a Pro-apoptotic as well as Anti-apoptotic modulator
Nitric oxide and cell signaling pahways in Mitochondrial-dependent apoptosis