Description of the protein and its gene.
Human Tau is encoded on chromosome 17q21.
Isoforms of Tau protein
| Clone inserts/repeats | Number of amino acids | MW (kDa) |
| htau40 2N4R | 441 | 45.9 |
| htau39 2N3R | 410 | 42.6 |
| htau34 1N4R | 412 | 43.0 |
| htau37 1N3R | 381 | 39.7 |
| htau24 0N4R | 383 | 40.0 |
| htau23 0N3R | 352 | 36.7 |
| big Tau 2N4R | 695 | 72.7 |
To date, the only established function of Tau is the promotion of assembly of tubulin into microtubules and stabilization of their structure. The protein occurs mainly in the axons of the CNS and consists largely of six isoforms generated by alternative splicing; the longest of them has 441 amino acids.
They differ in containing three (3R Taus) of four (4R Taus) microtubule binding repeats of 31-32 amino acids each in the carboxy terminal half and one (1N), two (2N) or zero (0N) amino terminal inserts of 29 amino acids each. The amino terminal fragment does not bind to microtubules but projects away from the microtubule surface and hence is termed "projection domain". The overall amino acid composition is usually hydrophilic, consistent with the unfolded character of the protein. In full lenght Tau there are 80 S or T residues, 56 negative (D+E), 58 positive (K+R), and 8 aromatic; so the protein has an overall basic character, but the amino terminal 120 residues are predominantly acidic and the carboxy terminal 40 residues are roughly neutral.
The middle region (AA 150-240) contains numerous prolines and also the region downstream of the repeat domain presents several prolines: these domains have acquired a special importance in the field because they harbor many epitopes of antibodies that become hyperphosphorylated in Alzheimer Disease, and are therefore diagnostic for the disease state.

Because of its hydrophilic nature Tau does not adopt a compact folded structure; in fact evidence suggests that the entire Tau molecule is "natively unfolded" or "intrinsically disordered". The polypeptide chain is highly flexible and mobile, there is only a low content of secondary structures, which are, however, transient. This corresponds to the observation that Tau can fulfill its physiological function of stabilizing microtubules even after harsh treatment (heat, acid,...).
Biochemistry and Cell Biology of Tau Protein in Neurofibrillary Degeneration 2012
Tau and microtubules.
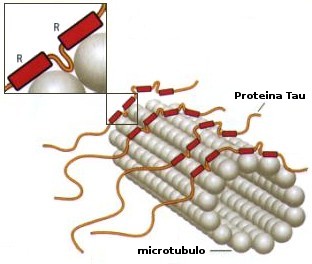
Tau and other MAPs stabilize microtubules by binding to the MT surface and promote their self-assembly from tubulin subunits. Reported dissociation constants vary from 0.02 to 1 microM, depending on isotype, mutations, phosphorylation and other modifications. The exact binding site of Tau is not known; the uncertainty is largely due to Tau's loose and natively unfolded structure. Recent studies have demonstrated very tight microtubule binding for the Tau fragment 208-304: this argues that the central part of Tau is aligned along protofilaments, consistent with cryo-electron microscopy data. In neurons, Tau is strongly substoichiometric compared with tubulin (20-40 microM tubulin compared with 1 microM Tau or less).The highly acidic carboxy terminal 30 residues of tubulin are important for tight binding, but they are also natively unfolded.
In considering functions of Tau towars microtubules, we can distinguish direct and indirect interactions. Direct interactions include the binding, stabilization and promotion of MT assembly that can be modulated by Tau and its phosphorylation. This function requires the MT binding domain (repeats + flanking domains), but not necessarily the projection domain. Another effect is the protection of microtubule ends against lenght fluctuations (dynamic instability), that is their ability to shrink and grow in stochastic manner.
Indirect interactions affect other proteins that may or may not interact with MT by themselves and may require the projection domain of Tau. An example is the spacer function of MAPs, which helps to establish a "clear zone" around microtubules in cells; another is the inhibition of Mt-dependent transport by motor proteins, which is based on the competition between motors (kinesin, dynein) and MAPs for binding sites on the MT surface. Finally, because MTs are distributed throughout cellular space, they (thanks to the MAPs) provide natural anchoring structures for cell components in general: this includes major brain kinases (e.g. Fyn, Src, Lck, Abl) that can bind to the PXXP motifs in the proline-rich domain of Tau through their SH3 domain.
Biochemistry and Cell Biology of Tau Protein in Neurofibrillary Degeneration 2012
Distribution of Tau.
The majority of Tau is located in the brain, specifically in neurons. It is ubiquitous in immature neurons but becomes axonal during maturation, accompanied by a shift toward higher molecular weight isoforms and reduced phosphorylation. Even after maturation low levels of Tau are present in other neuronal compartments, like the nucleus and dendrites, and in other brain cells, notably oligodendrocytes. Moreover, Tau mRNA can be detected in many cell types, even in muscle fibers where Tau forms aggregates in inclusion body myositis. The pathway of neuronal sorting is still uncompletely understood; transport of Tau into axons occurs as part of the slow axonal transport component SCa, probably as part of a (transient) complex with tubulin oligomers driven by kinesin.
The issue of axonal sorting is important because misassorting of Tau into the somatodendritic compartment is recognized as one of the earliest signs of neurodegeneration in AD.
Entry of Tau into dendrites (e.g. triggered by A-beta, glutamate, oxidative stress,...) causes a decay of dendritic spines mediated by Calcium influx through NMDA receptors and thus a decay of neuronal communication. Tau is not merely transported as a cargo but can actively interfere with micro-tubule dependent traffic because it can compete with motors for binding to microtubules and thereby slow down both anterograde and retrograde transport.
Tau is normally a cytosolic protein, but its occurrence outside cells has become important for studying and diagnosing the AD disease process. One reason is that Tau is elevated in the cerebrospinal fluid at an early stage of AD and therefore serves as a biomarker. The origin of CSF-Tau is not clear, but dying and disintegrating neurons could contribute to it.
Another recent development is derived by the observation that Tau pathology spreads in the brain in a well-defined manner, so that its distribution can be correlated with the clinical stages of disease. One hypothesis is that affected neurons release pathological Tau, which is taken up by neighboring cells and thus spreads the pathology in a prion-like fashion.
Biochemistry and Cell Biology of Tau Protein in Neurofibrillary Degeneration 2012
Tau binding partners.
Since Tau is very flexible and carries many charges it has the capacity to interact with many partners. By far the most important interaction of Tau is that with microtubules, but also interactions with other cytoskeletal elements have been reported as well, notably intermediate filaments and microfilaments. These non-MT interactions need not be direct, but could be mediated by other cytoskeleton-associated proteins. Binding sites of Tau have been reported for several kinases and phosphatases; the best-defined kinase interaction sites are the PXXP motifs in the proline-rich domain that can bind to SH3-containing proteins including tyrosine kinases Fyn, Src or Lck. A number of studies have dealt with the interactions of Tau with the chaperone system. Proteins such as hsp70 and their binding partners are thought to control the level of soluble Tau and thus help to prevent aggregation in a phosphorylation-dependent manner. Among the non-protein interaction partners of Tau, the interactions with the plasma membrane and with nuclear components have been intriguing over years. The emerging picture is that a small fraction of cellular Tau binds to the membrane through membrane-associated proteins, for example annexins or proteins of lipid rafts.
Formation and structure of Tau fibers.

Given that Tau is hydrophilic, unstuctured and dynamic, its aggregation into seemingly well-ordered and periodic fibers in AD is counterintuitive. However, two factors contribute to pushing Tau toward aggregation. One is the charge compensation of the basic middle part of Tau by polyanions. In vitro this can be achieved by sulfated glycosaminoglycans, while in neurons the inducers of Tau fibrillization are not known (but acidic cofactors are likely). Since the Tau-Tau interaction site largely overlaps with the Tau-MT interaction site, it appears that MTs override Tau's capacity for fibrillization, perhaps by stabilizing a nonaggregant conformation. Thus, MTs act effectively as chaperones for Tau to prevent their abnormal aggregation.
The second factor essential for Tau assembly is its propensity for beta structure, encoded in short hexapeptide motifs at the beginning of R2 and R3: disruption of these motifs abrogates Tau's tendency to aggregate, not only in vitro, but also in cell and animal models. While the presence of a cross-beta structure in the core of PHF is unambigous, its arrangement in detail is still unknown. Electron paramagnetic resonance spectroscopy studies suggests that the initial 20 residues of R2 and R3 are stacked axially in the extended beta-structure.
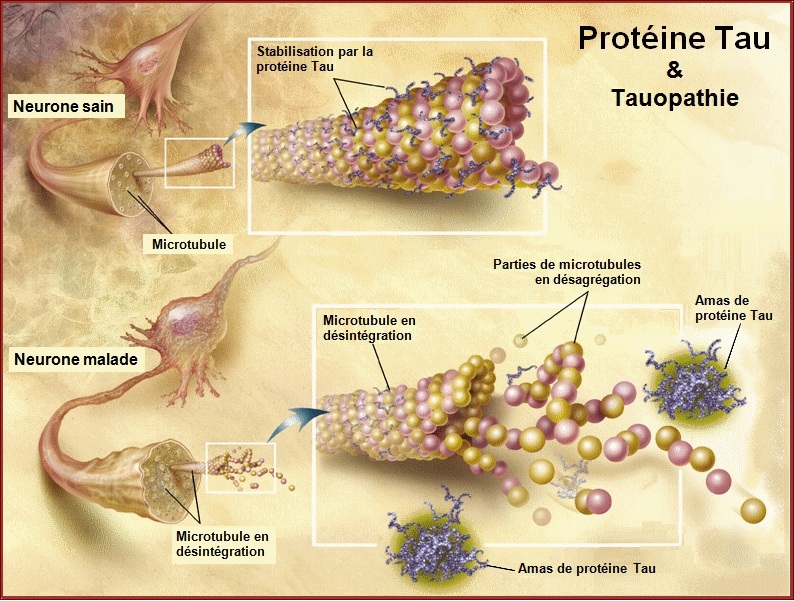
Pathological forms and states of Tau.
Neurofibrillary degeneration of abnormally hyperphosphorylated Tau not only occurs in AD brain but is also seen in a family of related neurodegenerative diseases, called tauopathies, such as frontotemporal dementia with Parkinsonism linked to chromosome 17 caused by Tau mutations, Pick's disease, corticobasal degeneration, dementia pugilistica and progressive supranuclear palsy.
In every one of this tauopathies the neurofibrillary changes are made up of abnormally hyperphosphorylated tau and their occurrence in the neocortex is associated with dementia.
In FTDP-17 several missense mutations in Tau cosegregate with the disease. In AD brain all of the six Tau isoforms are hyperphosphorylated and aggregated into PHF.
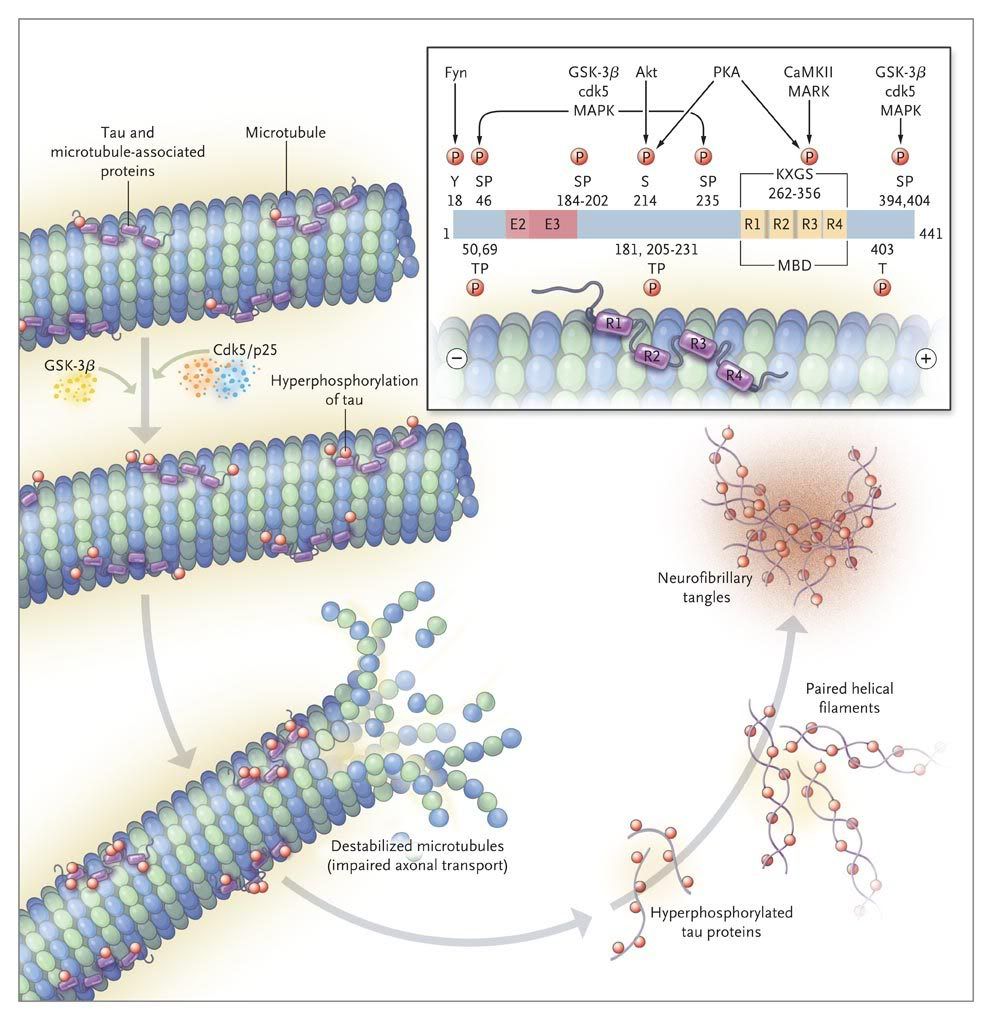
While in the normal brain almost all tau is soluble, from AD brain this protein is recovered in three major states, i.e. soluble, oligomeric and fibrillized. There is at least as much normal cytosolic Tau in AD brain as in normal aged brain but the level of total Tau in the former is four to eight fold higher and this increase is solely in the form of the abnormally hyperphosphorylated protein. As much as 40 % of the abnormally hyperphosphorylated Tau in AD brain is present in the cytosol and not polymerized into paired helical filaments/neurofibrillary tangles. The AD cytosolic abnormally hyperphosphorylated Tau (AD P-Tau) does not bind to tubulin and promote microtubule assembly, but instead it inhibits assembly and disrupts microtubules. This toxic property of the pathological Tau involves the sequestration of normal Tau by the diseased protein. The AD P-Tau also sequesters the other two major neuronal microtubule associated proteins MAP1A/B and MAP2. This toxic behavior of the AD P-Tau appears to be solely due to its abnormal phosphorylation because dephosphorylation of diseased Tau converts it into a normal-like protein. In wild-type human Tau and mutated human Tau transgenic Drosophila, the accumulation of the AD P-Tau in the absence of its fibrillization into neurofibrillary tangles leads to neurodegeneration: this suggests that the accumulation of the cytosolically AD P-Tau, and not its aggregation, is apparently involved in behavioral impairment. Furthermore, in vitro dephosphorylation of neurofibrillary tangles disaggregates filaments and, as a result, the Tau released behaves like normal protein in promoting microtubule assembly.
Thus, two characteristics of AD P-Tau are that it sequesters normal MAPs and disrupts microtubules and that it self-assembles into PHF.
In a recent study methyltioninium chloride (methilene blue dye) has been found to disaggregate PHF in vitro, reduce the number of Tau aggregates in Tau transgenic mices and show significant inhibition of cognitive impairment in a PHASE II double blind clinical trial in AD patients.
It is however worth to underline that hyperphosphorylation of Tau, though not on the same level as in AD, is not only associated with disease, but is also employed by the neuron to down regulate its activity transiently and reversibly when required.
Both the amino terminal and the carboxy terminal flanking regions to the microtubule binding repeats in normal Tau appear to inhibit its self-aggregation into filaments and on AD type abnormal hyperphosphorylation, i.e. the phosphorylation of the amino terminal and the carboxy terminal flanking regions, this inhibition is eliminated, resulting in the formation of tangles of PHF.
In addition to abnormal hyperphosphorylation, conformational changes and cleavage of Tau have also been implicated in the pathogenesis of AD. The hyperphosphorylation of Tau has been found to precede both conformational changes and cleavage of this protein. Tau contains many potential cleavage sites accessible to multiple proteases, yelding breakdown products that could be toxic in various ways. For example cleavage of the tails by caspases perturbs the paperclip folding of Tau and makes it more vulnerable to aggregation.
Other biochemical modifications are oxidation, ubiquitination, O-GlcNac glycosylation, acetylation (which impairs MT binding of Tau, promotes aggregation and potentially interferes with ubiquitination and thus degradation of Tau).
Finally, because Tau and its aggregates are long-lived, they are subject to nonenzymatic modifications. One is glycation by AGEs (advanced glycation end products); this decreases MT binding, promotes aggregation, activates RAGE receptors and thus causes oxidative damage. The increased level of Tau in AD brain is probably mostly due to a decrease in its turnover caused by the hyperphosphorylation. The AD P-Tau is resistant to both Calcium activated neutral proteases, calpains, and its degradation by the ubiquitin-proteasome pathway.

The many faces of Tau 2011
Treatments targeting Tau.
Treatments targeting various aspects of Tau biology are under intense investigation. Inhibitors of Tau phosphorylation and aggregation and microtubule stabilizers are already in clinical trials for people with AD or FTLD, while Tau reduction strategies are still in preclinical stages of development.
As we learn more about how Tau expression is regulated and about Tau's involvement in cell signalling and cytoskeletal organization, addiotional approaches are likely to emerge.
Tau in Alzheimer Disease and Related Tauopathies 2010