Authors: Andrea Saglietto, Martina Petitti
DEFINITION
The CONGENITAL CENTRAL HYPOVENTILATION SYNDROME (CCHS) is a disorder characterized by hypoventilation and hypoxemia. It appears early in life and is not associated with cardiopulmonary or neuromuscular abnormalities.
In 2003, the disease-causing gene for CCHS was discovered in the paired-like homeobox gene PHOX2B.
CCHS is also known as Ondine’s curse. This name comes from mythology: according to French and German mythology, the nymph Ondine discovered that her husband had committed adultery. Because he had promised his every waking breath to her, she cursed him that so long as he was awake he could breathe, but if he ever fell asleep he would stop breathing and die.

EPIDEMIOLOGY
Prevalence
Nearly 1,000 children worldwide have PHOX2B mutation-confirmed congenital central hypoventilation syndrome. However, some feel that this number is likely underestimated, due to the introduction of more extensive screening measures for PHOX2B mutations.
Incidence
Congenital central hypoventilation syndrome (CCHS) is a very rare disorder, with an estimated prevalence of 1 case per 200,000 live births. However, the introduction of more extensive screening measures for PHOX2B mutations has revealed that CCHS is not as rare as previously considered. Current estimates are likely an underestimate.
Sex
Both sexes appear to be equally affected.
Age
CCHS is present at birth, although the diagnosis may be delayed because of variations in the severity of the manifestations or lack of awareness in the medical community, particularly in milder cases. Late-onset CCHS may present in the school-aged child to adult years following an inciting infection or surgical procedure.
( Medscape, CCHS epidemiology )
SYMPTOMS
Patients suffer primarly from respiratory defect, that represents the most important evidence of the disease: this defect is due to a defective autonomic control of respiration and the absence of the normal response to hypercapnia ( Phox2b, congenital central hypoventilation syndrome and the control of respiration, 2013 ).
Looking at the figure ( Congenital central hypoventilation syndrome in children, 2011 ), we can see well the respiratory defect:
- In figure C, a portion of a polysomnogram from an infant with congenital central hypoventilation syndrome. In the left panel, the infant is awake with normal oxyhemoglobin saturation but is hypoventilating slightly. At sleep onset (middle panel), end-tidal carbon dioxide levels begin to rise and oxyhemoglobin levels begin to drop. When placed on supplemental oxygen (right panel), the oxyhemoglobin levels normalize but end-tidal carbon dioxide levels continue to rise.
- In figure D, a polysomnogram portion from an 8 year old subject with CCHS during NREM sleep is shown. Following ventilator disconnection (arrow), the subject had an immediate 24 second central apnoea, followed by an arousal.

Respiratory disfunction may range from mild hypoventilation during sleep whit adequate ventilation during wakefulness to complete apnea during sleep and severe hypoventilation when awake. This variability can be explained by the fact that the disease is caused by a mutation of Phox2b, a transcription factor expressed in a limited set of central and peripheral neurons. Depending on whether the mutation interests both alleles or not, the phenotype is different.
Patients can also show other possible sympthoms relating to SNA such as:
- Hirschsprung disease
- a propensity to develop neuroblastomas
- dysphagia
- reduced heart rate variability
- increased incidence of cardiac arrhythmia.
In addiction to all these sympthoms, some studies conducted by Todd and colleagues ( Congenital Central Hypoventilation Syndrome and the PHOX2B Gene Mutation, 2011 ) describe the existence of a specific facial phenotype in individuals with CCHS: faces are shorter and flatter, with decreased upper-face height, excessive nasal tip protusion, decreased nasolabial angle, short upper-lip height and an inferior inflection of the lateral segment of vermillion border on the upper lip.
PATHOPHYSIOLOGY
The control of breathing
Breathing is a rhythmic motor behaviour driven by a central pattern generator (CPG) located in the ventral medulla, which feeds onto the motor neurons that innervate respiratory muscles. This respiratory neurons are distributed in a ventrolateral column extending from the facial nucleus (nVII) to the spinal cord and subdivided into compartments that differ in connectivity and function. The best-characterized component of the CPG is the pre-Bötzinger complex (preBötC), whose oscillatory activity entrains and is an essential source of respiratory rhythm.
There is also a small group of neurons in the rostro-ventral medulla that represents an essential site for breathing because it’s responsible both for a normal respiratory rhythm in the fetal and neonatal period and for the tonic drive afforded by pCO2 throughout life. Depending on the developmental stage and its electrophysiological properties, this group of cells has been termed in different ways although they are a single entity: retrotrapezoid nucleus (RTN) in the adulty, parafacial respiratory group (pFRG) in the neonat, or embryonic parafacialgroup (e-pF) in the fetus.

The general features of RTN are:
- The homeobox transcription factor Phox2b is essential for the development of RTN neurons and, in general, for the development of respiratory network
- It has been demonstrated that the RTN area contains neurons activated by CO2 in the brain, due to connections with central chemoceptors located in the ventral surface of medulla, near respiratory centers. Moreover, they are likely able to sense pCO2 level directly by themselves.
However it's important to underline that RTN neurons are not only activated by pCO2 in the brain but also receive potent excitatory inputs from the carotic bodies and the aortic bodies, increasing its role of chemosensory integration centre.
To understand how the pCO2 level is sensed, we have to analyse separately peripheral chemoceptor and central chemoceptors.
For what concerns peripheral chemoceptors the mechanism is the one that we can see in the following picture: inside the peripheral chemoceptor, CO2 reacts with H20 (the reaction is catalyzed by carbonic anidrase), with the resulting formation of HCO3- and H+. The rise in H+ concentration inhibits K+ channels, leading to cellular depolarization and secretion of neurotransmitter, which is dopamine. Dopamine excites afferent branches of CN IX, for carotid bodies, and CN X, for aortic bodies. These cranial nerves finally send information to medullary centers. It's important to underline that peripheral chemoceptors can also sense a decrease in pO2 level.

In central chemoceptors, which are only able to sense pCO2 level, the mechanism is different: CO2 can easily pass through blood brain barrier (BBB), and it is believed that CO2 can act directly on chemoceptor, just like in the peripheral chemoceptor, or it can acts indirectly through the H+ ions deriving from CO2 idration in CNS.

( Phox2b, congenital central hypoventilation syndrome and the control of respiration, 2013 )
PHOX2B: the gene and the protein
Hints toward the familiality of CCHS emerged between the 1980s and 2001.
In 2003, PHOX2B was found to be the disease-defining gene for CCHS. PHOX2B encodes a highly conserved homeo-domain transcription factor known to play a key role in the development of ANS reflex circuits in mice. PHOX2B protein is 314 aminoacids long. The homeodomain region spanning exons 1 and 2 (from residue 99 to residue 148) is responsible for the binding of this transcription factor to target DNA elements. In exon 3 there are two sequences characterized by stretches of 9 and 20 alanine residues, encoded by GC triplets. The stretche of 20 alanines is the relevant one, as we will explain below. The role of the other stretche is still unknown ( PHOX2B, Atlas of Genetics and Cytogenetics in Oncology and Haematology ).

Protein Aminoacids Percentage

Transcription factor PHOX2B
Comparing PHOX2B aminoacids percentage with another transcription factor which acts togheter with PHOX2B to enhance PHOX2A expression (see next paragraph), we can appreciate how there is a certain similarity between the two of them:

Transcription factor Sp1
Cellular functions
The DNA-associated protein encoded by PHOX2B gene is a member of the paired family of homeobox proteins localized to the nucleus. The protein functions as a transcription factor involved in the development of several major noradrenergic neuron populations and the determination of neurotransmitter phenotype. The gene product is linked to enhancement of second messenger-mediated activation of the dopamine beta-hydroylase, c-fos promoters and several enhancers, including cyclic amp-response element and serum-response element. ( Gene, NCBI )
Dopamine beta-hydroylase is the enzyme catalyzing the last step of noradrenaline synthesis, because it converts dopamine to noradrenaline:
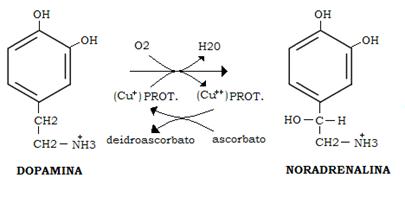
Besides both Phox2a and Phox2b are homeodomain proteins that are precociously and very similarly expressed in the three peripheral divisions of the autonomic nervous system. They are also expressed in all of the adrenergic and noradrenergic neurons of the brainstem, in some cranial sensory ganglia that participate in the autonomic reflexes, and in a subset of cranial motor neurons. Some studies have also demonstrated that, with the exception of the cranial sensory ganglia, Phox2a acts downstream of Phox2b and Mash1, a basic helix-loop-helix transcription factor that has been shown to promote competence for neurogenesis in neural crest progenitors. Together with Phox2b, Phox2a seems to act upstream of Ret (a subunit of the glia-derived neurotrophic factor receptor required for the survival of aggregating sympathoblasts and migrating enteric neuroblasts), and dopamine-β-hydroxylase, the last enzyme in the noradrenaline synthesis pathway as we have already discussed. ( SP Proteins and PHOX2B Regulate the Expression of the HumanPHOX2a Gene, 2001 )

Phox2b mutations
We can subdivide PHOX2B mutations into two groups:
- Polyalanine repeat expansion mutations ( PARMs ): it has been demonstrated that most of CCHS cases (90%) are heterozygous for an in-frame polyalanine repeat expansion mutations in the second stretche of alanine, coding for 24 to 33 alanines in the mutated protein and producing genotypes of 20/24 to 20/33 (the normal genotype would be referred to as 20/20).
- Non polyalanine repeat expansion mutations ( NPARMs ): the remaining approximately 10% of patients with a classical CCHS phenotype are heterozygous for an NPARM (including missense, nonsense, and frameshift) in the PHOX2B gene: individuals with CCHS and NPARMs in PHOX2B have been described worldwide, and mutations include predominantly frameshift mutations (78%), but also non-sense (4%), missense (16%), and missense with stop codon alteration (3%) The majority of CCHS-associated NPARMs are found at the end of exon 2 or in exon 3

NPARMs lead to a more severe respiratory phenotype, as well as very frequently they correlate also with Hirschsprung disease and with tumors of the neural crest cells, for example neuroblastoma:

( An Official ATS Clinical Policy Statement: Congenital Central Hypoventilation Syndrome, 2010 )
Biological differences between PARMs and NPARMs mutations of PHOX2B
PHOX2B mutations have been tested for potential disruption of the normal function of the protein with respect to (i) transactivation of different target promoters, (ii) DNA binding, (iii) aggregate formation, and (iv) subcellular localization. Distinct CCHS pathogenetic mechanisms for PARMs and NPARMs in PHOX2B have thus been postulated:
- PARMs: to investigate how PHOX2B PA expansions can induce CCHS pathogenesis, the ability of expression constructs containing PA mutations to regulate the transcription of known target genes has been compared, in two different laboratories, to a wild-type PHOX2B construct. In particular, as exemplified in Figure A for the DbH target gene, when mutant PHOX2B constructs were cotransfected with the DbH and PHOX2A regulatory regions connected to the Luciferase gene, a strict inverse correlation between the induced Luciferase activity and the length of the PA tract was identified. This suggested that the transcriptional regulation of these two genes is directly dependent on the correct structure of the PHOX2B domain, including the 20-alanine tract and that longer tracts increasingly disrupt transcription.
Fluorescence microscopy of COS-7 cells expressing PHOX2B proteins fused to a green fluorescent molecule has shown that the wild-type PHOX2B protein is present almost exclusively in the nucleus. However, increasing length of the PA repeat does induce an increasing percentage of PHOX2B protein within cells to mislocalize to the cytoplasm.
As such, similar experiments performed in HeLa cells induced formation of PHOX2B polyalanine aggregates, although in different amounts compared with that observed in COS-7 cells, suggesting that mislocalization of the mutant protein is a common pathogenetic mechanism leading to impaired transcriptional activity of mutant PHOX2B containing aggregation-prone expanded PA tracts.
In the attempt to assess the fate of cells expressing PA- expanded PHOX2B, in vitro experiments have demonstrated that activation of the heat-shock response by the drug geldanamycin, a naturally occurring antibiotic, is efficient both in preventing formation and in inducing the clearance of PHOX2B preformed PA aggregates and, ultimately, also in rescuing the PHOX2B ability to transactivate the DbH promoter. In addition, elimination of PHOX2B mutant proteins by the proteasome and autophagy, two cellular mechanisms already known to be involved in the clearance of proteina containing expanded polyglutamine and polyalanine tracts, has been demonstrated. Cellular apoptosis has been observed only in association with the largest PA expansions.
- NPARMs: Non-PA mutant PHOX2B proteins tested so far have shown compromised transcriptional activation of the DbH and TLX2 promoters with more severe activity disruption correlated with length of the frameshifted C-terminal sequence (see figure B for the effect on the DbH target). Unexpectedly, PHOX2B frameshift mutations have shown a 10 to 30% increased activation of the PHOX2A regulatory region. Moreover, frameshifts and missense mutations have mainly shown a complete loss of DNA binding but, unlike the long PA expansions, are able to correctly localize in the nucleus.
Aberrant C-terminal regions may cause PHOX2B protein dysfunction due to either lack of ability to establish correct protein–protein interactions with molecular partners or gaining of the ability to interact with wrong molecules, a very attractive hypothesis in light of the association of NPARM mutations with risk of neuroblastoma development. Consistently, a recent study has shown that non-PA mutant PHOX2B constructs retained the ability to suppress cellular proliferation without being able to promote differentiation, suggesting a mechanism that might promote the development of neural crest tumors.

In conclusion, these studies have indicated a marked difference between PARMs and frameshift NPARMs in terms of transactivation of target promoters, formation of aggregates, and subcellular localization. Future in vitro investigation will provide further clues on pathogenesis and possible therapeutic hints.
( An Official ATS Clinical Policy Statement: Congenital Central Hypoventilation Syndrome, 2010 )
A possible explanation of the unresponsivness to hypercapnia due to PHOX2B impaired activity
Now we have all the elements for trying to understand what happens in patients with Ondine's curse. In literature we cannot find anything certain about what is the real mechanism by which an impaired activity of PHOX2B results in lack of increased CO2 response, with consequent problems in breathing during sleep, the time when the chemoceptive trigger of the ventilation becomes the most important one.
Our hypothesis is the following: in CCHS patients the problem is not the fact that they don't sense "high pCO2", because central chemoceptors (except for RTN neurons, but there are other central chemoceptors) and peripheral chemoceptors of carotic bodies and aortic bodies work properly and their firing constitutes a great stimulus for the respiratory network to sustain more ventilatory effort. In few words, the afferent signals of an increase in pCO2 are properly generated and sent to respiratory centers.
The real problems stand in the efferent response of the respiratory centers. These problems are caused by PHOX2B in two possible ways:
- Defects in PHOX2B result in a selective impaired development of RTN nucleus, so that the efferent response to hypercapnia is greatly decreased. This is mainly due to decreased transcription and following expression of Ret, a subunit of the glia-derived neurotrophic factor receptor required for the survival of neurons
- Defects in PHOX2B result in a decreased transcription of dopamine-β-hydroxylase, which is the enzyme catalyzing the last step of noradrenalin syntesis. RTN neurons are glutamatergic, but a lot of other respiratory centers involved in respiratory reflex use noradrenaline as neurotrasmitter, such as A5 cell group, NTS and locus coeruleus. So, these neurons, involved in respiratory network, has a greatly reduced pool of their proper neurotrasmitter. This means that even if the afferent signals arrive correctly to them, these neurons will have a reduced secretion of neurotrasmitter, resulting in a defective response to hypercapnia.

DIAGNOSIS
CCHS is diagnosed in individuals with the following:
- Hypoventilation with absent or attenuated ventilatory response to hypercapnia and/or hypoxemia
- Generally adequate ventilation while awake and at rest and apparent hypoventilation with monotonous respiratory rate and shallow breathing (diminished tidal volume) during sleep OR apparent hypoventilation while both awake and asleep
- Absent perception of asphyxia (i.e., absent behavioral awareness of hypercapnia and/or hypoxemia) and absent arousal from sleep with development of physiologic compromise secondary to hypercapnia and/or hypoxemia
- No evidence of primary neuromuscular, lung, or cardiac disease or identifiable brain stem lesion that could account for the full constellation of signs and symptoms including autonomic nervous system dysregulation (ANSD)
- Presence of a CCHS-related PHOX2B mutation
- Symptoms of ANSD including but not limited to severe breath-holding spells; lack of physiologic responsiveness to the challenges of exercise and environmental stressors; diminished pupillary light response; esophageal dysmotility; severe constipation even in the absence of Hirschsprung disease; profuse sweating; reduced basal body temperature; and altered perception of anxiety
( PubMed books: Gene Reviews )
THERAPY
No medication has been shown to be effective in the treatment of CCHS, but some treatments exist and they allow patients to survive. They are:
- Invasive mechanical ventilatory support:
Positive-pressure ventilation through a permanent tracheostomy. Especially for children with CCHS.
Pressure plateau ventilation is suggested to be a useful alternative in home mechanical ventilation of children with CCHS who are unable to be adequately ventilated by standard volume control. The pressure-limited plateau ventilatory technique uses a volume ventilator but in a pressure-limit assist/control mode. Therefore, large volumes can be dialed as tidal volume into the ventilator. Because pressure is limited, the excess volume, which was not used to achieve the pressure plateau with each breath, is discarded via the pressure relief valve. This approach allows for breath-by-breath compensation of variable leaks.
- Noninvasive ventilatory support:
Negative-pressure ventilation in CCHS patients.
The tool most used in this method is the nasal mask (called bi-pap). It’s very effective in patients with CCHS who are older than 7-8 years and who are nocturnally dependent on the ventilator.
However, this method is not used much because of its side effects, such as upper airway obstruction during sleep in younger children with CCHS, and its equipment adjustments requests over time.
- Diaphragm pacing:
It regularly emits electrical impulses that allow the diaphragm to move and make breathing the patient.
Diaphragm pacer settings must provide adequate alveolar ventilation and oxygenation during rest and daily activities. Long-term outcome appears good, especially quality of life.
- Surgical cares:
Tracheotomy for ventilatory support and diaphragmatic pacing in appropriate patients (check the points above).
Today researchers are conducting some studies to find medical therapies about CCHS. One report of recovery with desogestrel is quite interesting: in vitro the antibiotic geldanamycin (GA) and its analog, 17-AAG, and curcumn, a compound found in turmeric spice, have been shown to promote clearance of mutant proteins resulting from expression of the PHOX2B gene with expanded polyA regions. Extending these investigations to in vivo studies is needed before they can be used in CCHS patients.
( Medscape, CCHS treatment )