DEFINITION
Danon Disease or Glycogen Storage Disease Type IIb is a genetic metabolic disorder causing hypertrophic cardiomyopathy. Mutations of the LAMP2 gene have been reported in association with this disease.
 Danon
Danon
LAMP2
INTRODUCTION
The patients, who were described in the January, 1981 issue of Neurology, were remarkably similar both clinically and pathologically. Both were 16-year-old boys, with proximal weakness, cardiomyopathy, and mild mental retardation since childhood. Both died at age 17 of cardiac failure. Their muscle biopsies were virtually identical and showed vacuoles reacting intensely both with the periodic acid Schiff (PAS) and with the acid phosphatase reactions, indicating that they were glycogen-laden lysosomes. Ultrastructural studies showed abundant glycogen particles, most of which were contained within lysosomal sacs, either alone or together with cellular debris; the vacuoles had the appearance of autophagic vacuoles. Although heart and brain are typically not affected in the juvenile form of acid maltase deficiency (AMD, glycogen storage disease type II), the pathological features of the muscle biopsy were typical of AMD. The finding of normal or higher-than-normal activities of AM (acid α-glucosidase) both with the artificial fluorogenic substrate and with natural substrates (glycogen and maltose) prompted to publish these cases as “Lysosomal glycogen storage disease with normal acid maltase”.
As more cases were described in the years that followed, it became clear that this was a unique and characteristic entity invariably involving lysosomes but with inconsistent glycogen storage, and clear X-linked inheritance.
EPIDEMIOLOGY
The incidence of Danon Disease has not been determined. HCM is estimated to be present in 2 of every 1000 young adults, according to one large study.
Charron et al. examined 197 independent index cases with HCM. Genomic sequencing for the LAMP2 gene revealed mutations in 2 of 197 (1%) patients with HCM.
Reports from several countries describe Danon disease in patients of several nationalities. However, the incidence of Danon disease appears to be too low to allow investigators to estimate its frequency in a given population.
The disease is inherited as an X-linked dominant trait, although spontaneous mutations have been reported in several families. Males are usually more severely affected than females, particularly in degree of cardiomyopathy and age of death. This difference is at least partially explained by a gene-dosage effect. Male individuals have only one X chromosome with the LAMP2 mutation, and most female individuals have one X chromosome with the mutation and one normal X chromosome; therefore, the effect is most pronounced in males.
The age of presentation is somewhat variable. The greatest factor that affects the age at presentation is the patient's sex. The age at presentation can range from infancy to the second decade in males but is less well defined in females. Male patients typically present in their teens and rarely survive beyond their 20s.
Lacoste-Collin et al reported a new diagnosis of Danon disease in a 41-year-old man.
SYMPTOMS
Danon disease usually manifests with the clinical triad of cardiomyopathy, skeletal myopathy, and mental retardation. Regardless of sex, cardiomyopathy can present as a result of symptoms or congestive heart failure (CHF) or an arrhythmia-related event, such as syncope or sudden death. Patients are also newly identified when asymptomatic relatives of patients with established Danon disease are evaluated and are found to have the disease.
Specific cardiac symptoms
- Male patients may present
- palpitations
- arrhythmias
- syncope
- chest pain
- cardiac arrest
The heart disease (cardiomyopathy) can be severe and can lead to a need for medications. It usually progress to heart failure, commonly complicated by atrial fibrillation and embolic strokes with severe neurological disability, leading to death unless heart transplant is performed.
Problems with the electrical conduction in the heart can occur. Sometimes the conduction problem is called Wolff-Parkinson-White syndrome.
- Female patients most typically present more with symptoms of dilated cardiomyopathy and CHF.
Specific neurologic symptoms
- weakness of the proximal extremities and neck muscles in the pattern of a limb-girdle muscular dystrophy.
- mental retardation or a learning disorder
- Some male patients present with neurologic symptoms in infancy, including difficulties in walking and delay in achieving developmental milestones
DIAGNOSIS
histopathology
Vacuolar myopathy is present with many vacuolar contents reacting positively with periodic acid-Schiff (PAS) stain and revealing increased acid phosphatase and nonspecific esterase activity. Normal architecture is seen on acetylcholine (ACH) stains without evidence of fiber grouping. No ragged red fibers are seen. Inflammation and fibrosis are absent. LAMP2 is absent on immunofluorescence or Western blots, whereas antibody stains for dystrophin and lysosomal-associated membrane protein-1 (LAMP1) are usually positive.
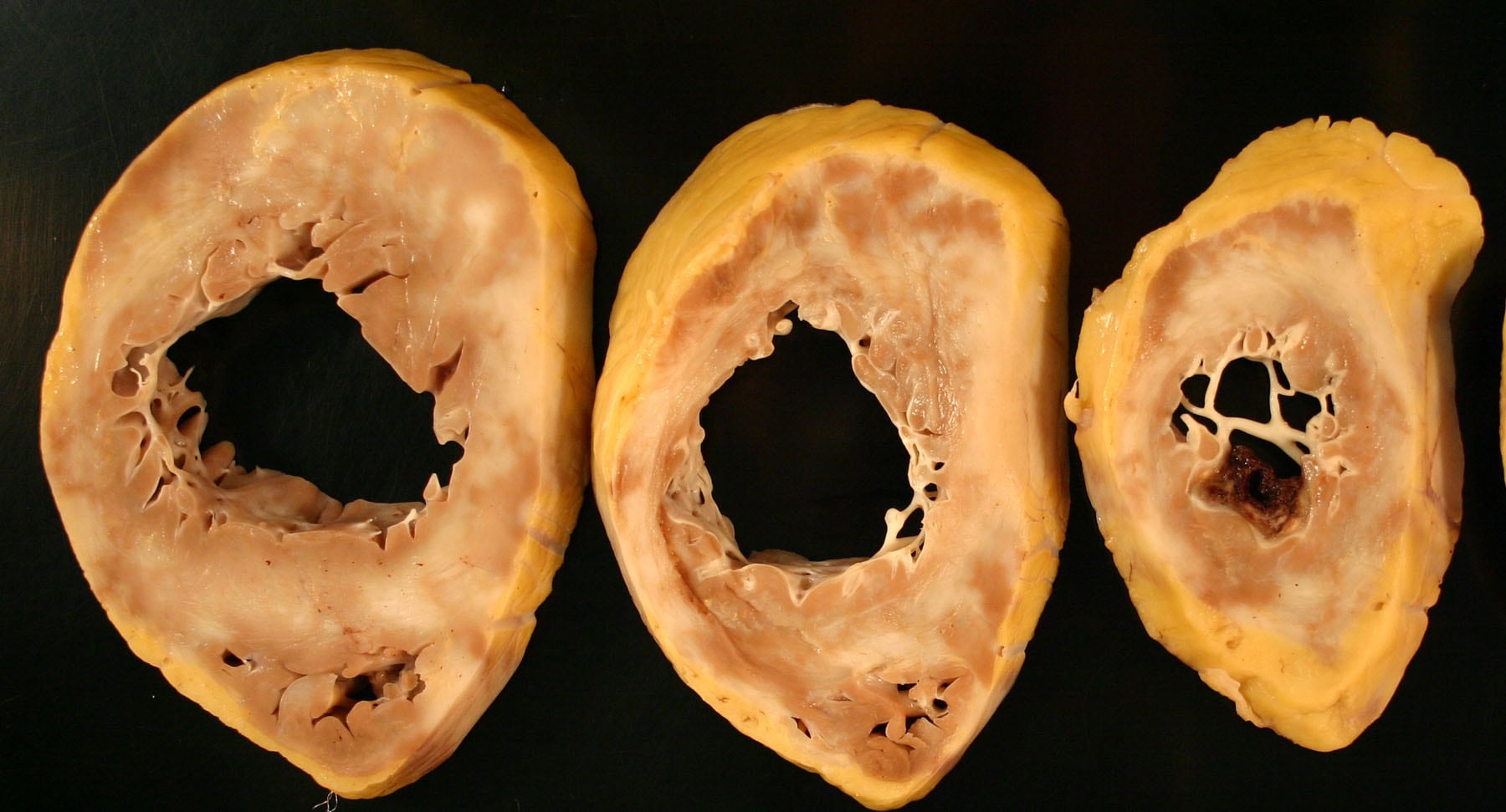
Horizontal ventricular sections of the heart from 16-year-old male adolescent with Danon disease obtained after orthotopic cardiac transplantation. Massive hypertrophy is present (heart weight, 785 g), with diffuse severe fibrosis and marked ventricular dilatation.
Electron microscopy reveals autophagic vacuoles and excess glycogen. The glycogen is both membrane bound and free between myofibrils.
Mitochondria have normal morphology.
Autopsy or explant specimens examined at the time of transplantation reveal cardiomegaly with ventricular hypertrophy and biatrial and biventricular dilatation. Interstitial fibrosis is often prominent, whereas myocardial vacuoles may not contain abundant PAS-positive material as commonly as is seen in biopsy material.
Echocardiography
Findings are abnormal in all patients with Danon disease
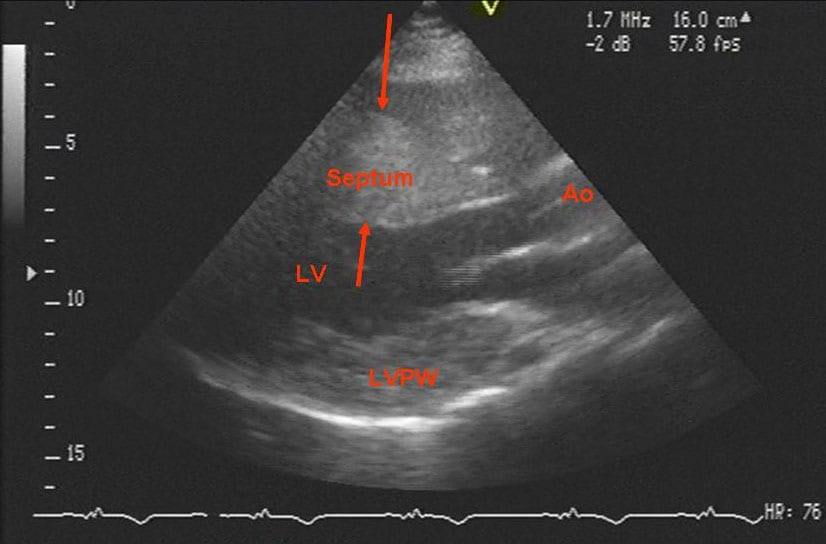
Echocardiogram of a patient with Danon disease and severe hypertrophy. The septum is between the arrows. Note the asymmetry between the septum and the posterior wall of the left ventricle. Ao = ascending aorta just above the aortic valve; LV = left ventricle; LVPW = left ventricular posterior wall.
MRI
- Cardiac MRI may be useful for assessing hypertrophy and function and to detect possible areas of poor gadolinium uptake that indicate scarring.
- MRI of the brain may reveal areas of involvement including hyperintensities of supratentorial white matter and cortical atrophy.
Molecular genetics studies
- Mutations in the LAMP2 gene were found in many patients with Danon disease.
- The LAMP2 gene is on chromosome Xq24 and contains 9 coding exons, with 2 alternate last exons: 9a and 9b.
- LAMP2a and LAMP2b are created with alternative splicing of exon 9a and 9b. LAMP2b is expressed most prominently in muscle and brain, and LAMP2a is expressed in greatest quantity in other tissues.
- Mutations in LAMP2 have included single or multiple base-pair deletions, additions, and substitutions that result in frameshift or nonsense mutations.
- Intronic mutations that produce skipping of one or more entire exons has been reported.

GeneLoc map region
around gene/marker "LAMP2"
Danon
Differential diagnoses
Protein Aminoacids Percentage
The Protein Aminoacids Percentage gives useful information on the local environment and the metabolic status of the cell (starvation, lack of essential AA, hypoxia).

Number of amino acids: 410
Molecular weight: 44963.02 = 44945.02 (+18)
LAMP2
mechanisms of fusion
LAMP-2 is a 410 amino acid protein consisting of a small cytoplasmic tail with a lysosomal membrane targeting signal, a transmembrane domain, and a large intraluminal head. The LAMP-2 open reading frame consists of 9 exons: the first 8 exons and part of the ninth encode the luminal domain, and what is left of exon 9 encodes both the transmembrane and the cytoplasmic domains. Human exon 9 exists in two forms, 9a and 9b, which are alternatively spliced, producing two isoforms, LAMP-2a and LAMP-2b. Nishino et al. provided evidence that Danon disease is mostly due to defects of the LAMP-2b isoform, which is predominantly expressed in heart, muscle, and brain, the three “target tissues” in Danon disease.

The schematic structure of the lysosome associated membrane protein LAMP-2
In autophagy, double-membrane vesicles called autophagosomes sequester part of the cytoplasm and then fuse with lysosomes to form hybrid-like organelles called autolysosomes. In S. cerevisiae, in which the molecular mechanisms of autophagy are best understood, autophagosomes fuse with the vacuole. The mechanism of fusion has similarities with homotypic vacuole fusion, SNAREs for which the likely mammalian orthologues are syntaxin-7 and VTI1B, have both been implicated in the fusion of autophagosomes with the yeast vacuole. In mammalian cells, RAB7 has been implicated in the fusion of autophagosomes with lysosomes. Fusion is reduced in cells that are depleted of LAMP1 and LAMP2, although the mechanism behind this effect is not understood. At least some other proteins that are required for autophagosome–lysosome fusion in mammalian cells are likely to be the same as those required for late endosome–lysosome fusion.

Delivery to lysosomes and lysosomal fusion.
Lysosomal membrane proteins LAMP1 and LAMP2 are transported from the trans-Golgi network (TGN) to lysosomes via clathrin-coated transport intermediates.
It is possible to hypothesize LAMP2's mechanism of action referring to Protein Aminoacids Percentage and sequence: evaluating these protein's localization, consequences to mutations and lack of LAMP-2, seems to be plausible that Lysosomes Associated Membrane Proteins are implicated in merging vesicles mechanism and fusion of autophagosomes with lysosomes through signal recognition of a specific amminoacid sequence.
LAMPs contain C-terminal tyrosine-based sorting signals YXXW (Y is tyrosine, X is any amino acid, and W is an amino acid with a bulky hydrophobic side chain (leucine, isoleucine, phenylalanine, methionine, valine), which bind the medium subunits of clathrin-adaptors (Ohno et al., 1998).
The position of YXXW signals within the tails of lysosomal integral membrane proteins might provide for optimal recognition at sites of sorting to lysosomes.
Another factor that is likely to affect signal recognition is the aggregation state of the integral membrane proteins. A considerable specificity overlap was also revealed by many analyses, suggesting that additional factors, such as the context of the signals, must be important determinants of recognition. For instance, aggregation caused by changes in the pH would be expected to increase the avidity of the proteins for membranebound adaptors.
PATHOGENESIS
The transmission of Danon disease is often X-linked dominant; however, spontaneous mutations have been documented. Phenotypic expression varies. The pathogenesis of Danon Disease is not well understood. It is attractive to assume that the clinical manifestations are the direct result of accumulation of glycogen in the lysosome, but this explanation is likely to be simplistic.

Pedigree of a family with Danon disease. Filled symbols indicate affected patients. The pattern of inheritance is compatible with X-linked dominant transmission, with male patients affected earlier and dying younger than hemizygous females.
A feature that distinguishes the vacuoles in Danon disease from typical lysosomes is that vacuolar membranes occasionally merged with indentations of the sarcolemma and stained with antibodies to sarcolemmal proteins, such as dystrophin and laminin. Based on the shared lysosomal and plasma membrane features of the vacuoles and on the X-linked inheritance of the disease, in 2000, Nishino and coworkers sequenced a candidate gene on chromosome Xq24, LAMP-2, in ten unrelated patients with Danon disease. They found pathogenic mutations in all patients and documented lack of LAMP-2 both by Western blot analysis and by immunohistochemistry. Their findings were bolstered by data from LAMP-2 knockout mice, which also showed accumulation of autophagic vacuoles in all tissues, but predominantly in cardiac and skeletal muscle.
Danon disease associated LAMP2 gene mutation results in a putatively truncated protein, which lacks the transmembrane domain and the cytosolic tail of the wild-type LAMP2. This variant becomes exocytosed because of a failure in targeting to late endosomes/lysosomes. Western blotting of cardiac muscle, White Blood Cells and cultured skin fibroblasts showed no intra- or extracellular truncated LAMP2. Comparing the expression pattern and intracellular targeting in cultured skin fibroblasts of normal LAMP2 isoforms (A, B and C) tagged with green fluorescent protein (GFP) , it results that the protein is not even expressed. These observations suggest that the truncated protein is unstable and is co-translationally or early post-translationally degraded.
The LAMP2 gene mutations that cause Danon disease lead to the production of very little or no LAMP-2 protein. Most mutations affect all three isoforms of the LAMP-2 protein. However, a mutation that affects only the LAMP-2B isoform also causes Danon disease, suggesting that the condition is caused by defects in the LAMP-2B protein.
Some studies have shown that in cells without the LAMP-2 protein, fusion between autophagic vacuoles and lysosomes occurs more slowly, which may lead to the accumulation of autophagic vacuoles. People with Danon disease have an abnormally large number of autophagic vacuoles in their heart and skeletal muscle cells. It is possible that this accumulation leads to breakdown of the muscle cells, causing the muscle weakness seen in Danon disease. Furthermore, it is unknown why some people with LAMP2 mutations develop hypertrophic cardiomyopathy but not the other features of Danon disease.
Serum and amino acid-starved LAMP2-negative cells exhibited an accumulation of autophagic vacuoles and then succumbed to cell death with hallmarks of apoptosis such as loss of the mitochondrial transmembrane potential, caspase activation and chromatin condensation. While caspase inhibition retarded cell death, it had no protective effect on mitochondria. Neither caspase inhibition nor mitochondrial stabilization antagonized autophagic vacuolization in LAMP2-deficient cells. Altogether, these data indicate that accumulation of autophagic vacuoles can precede apoptotic cell death.
COMPLICATIONS
ventricular septal thickness more than 30 mm is considered a risk factor for a life-threatening event
Potential complications for this disease include syncope or sudden death.
As with other forms of dilated cardiomyopathy, low-flow states can pose a risk of intracardiac thrombus formation with the potential for stroke.
THERAPY
Patients with Danon disease require frequent follow-up, with particular attention to the potential for atrial or ventricular arrhythmias and congestive heart failure (CHF).
Several surgical interventions should be considered in patients with Danon disease:
Beta blockers: These agents are indicated for the management of dilated cardiomyopathy.
ACE inhibitors: These agents reduce afterload in dilated cardiomyopathy and/or CHF.