Vitamin K
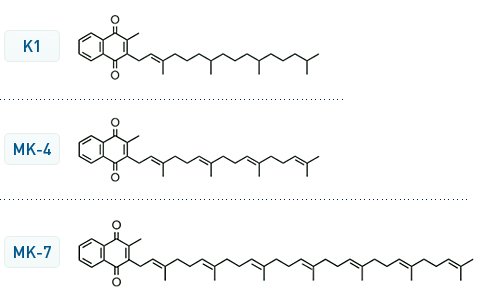
there are different forms of Vitamin K. The K1 is the usual one
Coenzyme Q10 in humans is used in the mitochondria to consume oxygen and produce energy. The left side of the molecules is very similar.
Coenzyme Q10 Structure


The main difference is the double methylation on the aromatic ring (Methylation is a feature of eukaryotic cells, not present in bacteria and virus??)

Vitamin K1 is used by aerobic bacteria in the intestine to consume oxygen and to produce energy.
that means that there is a competition between man and his intestinal bacteria for oxygen.
How a man can protect its own oxygen from the Vitamin K-bacteria?
Increasing blood clotting and closing vessels to avoid spreading of bacteria in the blood.
Clotting
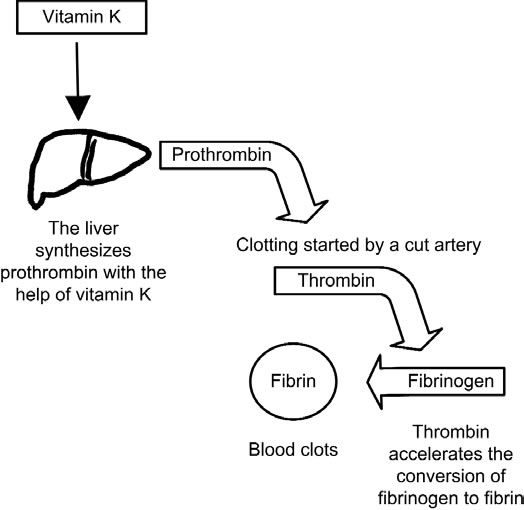
The more the aerobic bacteria, the more the Vitamin K, the more the clotting.
Too many aerobes in the gut too much clotting--> Thrombosis, heart infarction, ictus
Too few aerobes in the gut too few clotting --> Bleeding
The number of aerobic bacteria depends on the diet, the use of antibiotics not only for medical uses but also for growing chicken, pigs and for food conservation.
Vitamin K1
Vitamin K -dependent protein carboxylation is a process that involves all tissues and regulates processes like coagulation, apoptosis, signal transduction and arterial calcification.
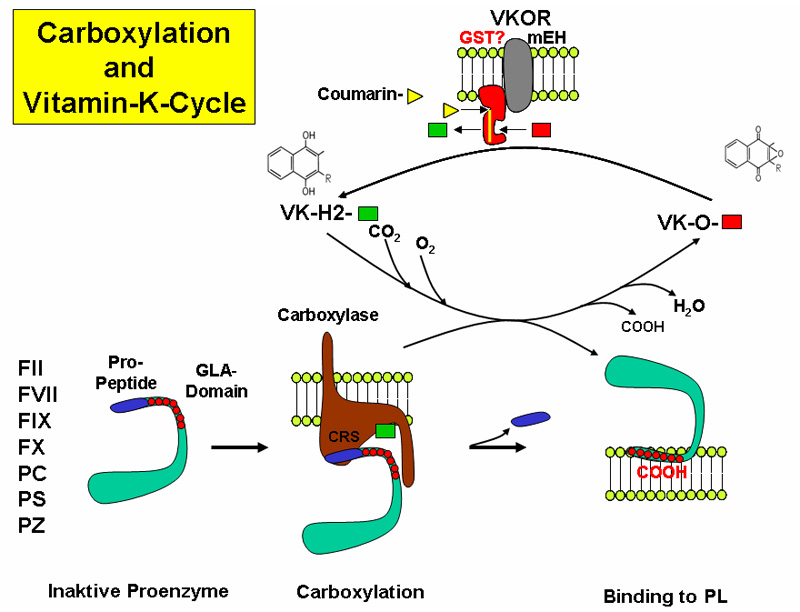
The reaction takes place in the endoplasmic reticulum leading to the formation of GLA domains, rich in gamma-carboxyglutamate (Gla), in the target proteins.

The reaction requires different steps:
- Vitamin K oxidation and protein reduction
- CO2 incorporation as carboxyl group
- recycling of the oxidized vitamin K back to the reduced form, by action of a Vitamin K Oxidoreductase (VKOR) that is the target of the anticoagulant drug Warfarin

Vitamin K Dependent (VKD) proteins
The Vitamin K–Dependent Carboxylase 2000
Synthesis of vitamin K-dependent proteins 1993
Google Books VKD proteins

VKD-γ-glutamyl carboxylase
Hypothetical model of the vitamin K-dependent γ-carboxylation system.
VKD-γ-glutamyl carboxylase activity regulation
Enhanced Renal Vitamin-K- Dependent Gamma-Glutamyl Carboxylase Activity in Experimental Rat Urolithiasis 1998
Open Questions
naturally-occurring human mutations in the carboxylase cause two distinctly different diseases
- pseudoxanthoma elasticum that is associated with dermal defects
- VKCFD1 (for combined deficiency of vitamin K-dependent coagulation factors) that is associated with serious bleeding.
The carboxylase is regulated by self-carboxylation
how the secretory machinery impacts carboxylation, which occurs in the endoplasmic reticulum during vitamin K-dependent protein secretion.
VKOR reduction of vitamin K may be the limiting step in carboxylation,
VKOR and the carboxylase, which are both integral membrane proteins, exist in a complex to cycle vitamin K ?
Roles of Vitamin K
Natural Vitamin K inibitors
Natural prenylquinones inhibit the enzymes of the vitamin K cycle in vitro. 1996
Quinones with a redox potential exceeding that of vitamin K1 (eg Ubiquinone10 = E(mv) 122) , showed strong inhibition of the KH 2 mediated carboxylation reaction, whereas the anthraquinone derivative (eg Menadione = E(mv) 2), with a relatively low redox potential, had no effect.
Fulltext
Vitamin K-antagonistic effect of plastoquinone and ubiquinone derivatives in vitro. 1994
Idem on Science Direct Fulltext
Vitamin K2
Vitamin K2 Is a Mitochondrial Electron Carrier That Rescues Pink1 Deficiency
Melissa Vos1,2, Giovanni Esposito1,2, Janaka N. Edirisinghe3, Sven Vilain1,2, Dominik M. Haddad1,2, Jan R. Slabbaert1,2, Stefanie Van Meensel1,2, Onno Schaap1,2, Bart De Strooper1,2, R. Meganathan3, Vanessa A. Morais1,2, Patrik Verstreken1,2,*
+ Author Affiliations
Vitamin K2 Is a Mitochondrial Electron Carrier That Rescues Pink1 Deficiency, 2012
ABSTRACT
Human UBIAD1 localizes to mitochondria and converts vitamin K1 to vitamin K2. Vitamin K2 is best known as a cofactor in blood coagulation, but in bacteria it is a membrane-bound electron carrier. Whether vitamin K2 exerts a similar carrier function in eukaryotic cells is unknown. We identified Drosophila UBIAD1/Heix as a modifier of pink1, a gene mutated in Parkinson’s disease that affects mitochondrial function. We found that vitamin K2 was necessary and sufficient to transfer electrons in Drosophila mitochondria. Heix mutants showed severe mitochondrial defects that were rescued by vitamin K2, and, similar to ubiquinone, vitamin K2 transferred electrons in Drosophila mitochondria, resulting in more efficient adenosine triphosphate (ATP) production. Thus, mitochondrial dysfunction was rescued by vitamin K2 that serves as a mitochondrial electron carrier, helping to maintain normal ATP production.
Vitamin K2 clinical use
vitamin+k2+cell+proliferation
vitamin+k+cell+proliferation
PROX1 suppresses vitamin K-induced transcriptional activity of Steroid and Xenobiotic, 2011
Recently, we have demonstrated that vitamin K2 also serves as a ligand for SXR and activation of SXR by vitamin K2 suppressed proliferation and motility of hepatocellular carcinoma (HCC) cells.
h3 Vitamin K liver efflux

The role of vitamin K in activation of gla-proteins. Vitamin K serves as a co-factor of activation of blood coagulation factors and matrix gla-protein (MGP) by γ-glutamyl carboxylase encoded by the GGCX gene. The γ-glutamyl carboxylase activates coagulation factors (Glu → Gla); the carboxylated forms are secreted into circulation from hepatocytes and are required for normal blood coagulation (lower panel). In peripheral connective tissue cells, such as fibroblasts, similar activation of uncarboxylated MGP (ucMGP) normally takes place resulting in carboxylated matrix gla-protein (cMGP), which is required for prevention of unwanted mineralization of peripheral tissues under normal calcium and phosphate homeostatic conditions (upper panel). It has been postulated that ABCC6 serves as a transporter molecule transporting vitamin K derivatives, such as the reduced form (KH2) conjugated with glutathione (GSH). In the absence of ABCC6 transporter activity in PXE, the specific vitamin K co-factor concentrations in the serum and in fibroblasts are reduced, resulting in deficient activation of MGP and allowing ectopic mineralization in the adjacent connective tissue to take place.

Common pathogenetic mechanisms in vascular calcification and osteoporosis.
BMPs participate in osteoblasts differentiation while they simultaneously produce ROS and increase the adhesiveness of monocytes on the vascular wall. Their action is blocked by MGP, a vitamin K-dependent protein, which also inhibits vascular mineralization as a co-factor of α-2-HS-glycoprotein (also known as fetuin-A). RANKL is a key factor of osteoclast maturation and also acts as an anticalcifying molecule. OPG prevents the interaction of RANKL with its receptor. Wnt signaling, which is important for osteoblast differentiation, is inhibited by sclerostin and DKK-1. On the vascular wall, Wnt is upregulated by the transcription factor MSX2, which blocks the inhibitory effect of DKK-1, resulting in increased vascular calcification. Phosphate, which penetrates the SMC wall through the Pit-1 co-transporter, directly stimulates vascular calcification whereas pyrophosphate acts as an inhibitor of calcification. OPN binds calcium and hydroxyapatite ions, thereby inhibiting crystal formation and vascular calcification; it interacts with integrin receptors resulting in osteoclast activation. PTH inhibits osteoblast activation and increases bone resorption; via PKA activation it induces osteoblastic differentiation and mineralization of vascular cells. Vitamin D increases the entry of calcium into vascular cells, resulting in calcification. Oxidized LDL cholesterol induces the expression of potent mediators of osteoclastic differentiation. Finally, angiotensin II participates in osteoclast activation. Abbreviations: BMP, bone morphogenetic protein; Ca2+, calcium; DKK-1, Dickkopf-1; E-NPP1, ectonucleotide pyrophosphatase/phosphodiesterase family member 1; M-CSF, macrophage-colony stimulating factor 1; MGP, matrix Gla protein; OPG, osteoprotegerin; OPN, osteopontin; Pi, inorganic phosphate; PPi, pyrophosphate; PKA, protein kinase A; PTH, parathyroid hormone; PTH1R, parathyroid hormone 1 receptor; RANKL, receptor activator of nuclear factor κB ligand; ROS, reactive oxygen species; RUNX2, runt-related transcription factor 2; SMC, smooth muscle cell; TRAP, tartrate-resistant acid phosphatase type 5.
Osteoporosis—a risk factor for cardiovascular disease?, 2012


The role of vitamin K2 in vascular calcification and osteoporosis
https://pubmed.ncbi.nlm.nih.gov/29880619/ Vitamin k by bacterial overgrowth
Weight control
The Effects of Vitamin K2 (Menaquinone-7) on the Leptin and Adiponectin Levels in Overweight/Obese Type 2 Diabetes Patients: A Randomized Clinical Trial, 2023