By Michela Nalin and Morena Trevisan
SSRI
The acronym SSRI (selective serotonin reuptake inhibitors) is to indicate, druggist molecules that re-enter within of selective inhibitors of the reuptake of the serotonin that is the so-called the not tricyclic antidepressants. Such drugs come used of norm in psychiatry for the therapy of pathologies which the obsessive-compulsive disturbance or the greater depression as, preventing to the normal reuptake and physiological elimination of the serotonin, they are in a position to contrasting eventual deficit of this neurotransmitter, re-balacing from the closely organic point of view the disturbances generated from its eventual deficiency.
The compounds belonging to the class of SSRIs are fluoxetine, a fluvoxamine, paroxetine, sertraline, the, the citalopram and recently introduced its active enantiomer, escitalopram. Specific characteristic of this group of drugs is the different affinity for the serotonin transporter. A distinctive feature of the SSRI is the lower incidence of side effects compared with tricyclic antidepressants (TCA), which is the direct consequence of the failure to take action at the level of receptors for various neurotransmitters.
At the level of serotonin transporter are two sites: a primary site, both of which bind antidepressant drugs blockers of serotonin transporter and a site of modulation allosteric. In the absence of inhibitors, serotonin is transported on a selective synaptic space within the terminal from which it was issued.
The SSRIs may also inhibit the oxidative metabolism of drugs metabolised by isoenzymes of the hepatic microsomal cytochrome P-450 (2D6, 1A2, 3A4, etc..), Increasing plasma concentrations, sometimes to a considerable extent. The degree of inhibition is dose-dependent. Exist between the SSRIs significant differences in the degree of inhibition exerted on the various isoenzymes. The most powerful drug to inhibit the CYP 2D6 was paroxetine, followed by fluoxetine and its metabolite norfluoxetine, sertraline also inhibits both in vitro and in vivo CYP2D6, with a capacity lower than paroxetine and fluoxetine. The potency of the SSRIs on the CYP2D6 is in the inhibit particularly emphasized because many drugs are metabolized by this enzyme.
The sertraline for instance is normally administered at doses that are 5-10 times higher than those of paroxetine and could result in a plasma concentration sufficiently high to compensate for the difference in inhibitory potency and provide the foundation for significant interactions with other drugs of the substrate CYP2D6.
Inhibition of the cytochrome P450 isoenzymes has important clinical implications given that the same enzyme metabolizes more than one drug. Therefore an interaction may occur when pharmacological antidepressants that inhibit a P450 isoenzymes are made simultaneously with drugs metabolized by that enzyme. Inhibition of cytochromes system may lead to a summation or synergy of the drug, increasing the effect of one or both drugs.
For example, the administration of a β-blocker and a lipophilic SSRIs that inhibit CYP2D6 can cause bradycardia as a result of increased plasma concentration of β-blocker. Although antidepressants are effective as monotherapy, are not uncommon associations and patients often switch from one drug to another in search of more effective treatment. In these cases there is a risk of an interaction if the elimination of the drug that was administered first is not complete.
The citalopram it is most selective between the SSRI, does not interact with other conveyors, it possesses a weak person affinity for the H-1 receivers of the histamine, neurochemical base of the light sedative effect, it does not inhibit the activity of CYP 450 and therefore it does not give place to dangerous interactions with other drugs.
The sertralina blocks also the conveyor of DA and this effect would above all explain its disinhibiting properties and noticeable activators to cognitive level in the old patients, but it could carry to an excessive stimulation of the patient, with anxiety manifestations.
The paroxetina inhibits also the uptake of the NA, inhibits the nitric oxyde synthase, effect that can carry to dysfunctions erected them, and turns out to be the molecule with greater anticholinergic action above all for an action at the level of the muscarinic receivers M3.
Such effect, besides the powerful inhibition of the CYP2D6, it demands a detail attention in the old patient with cognitive problems and in the polypharmacotherapy with other drugs metabolize from the same one isozyme.
The fluvoxamina, between the SSRI, turns out the molecule with mainly sedative characteristics sure; it is a powerful inhibitor of the isoform 1A2 and 3A4 of the CYP450 and therefore it can give place to interactions with other drugs that follow the same metabolic way. The fluoxetina inhibits also the uptake of the NA and this effect associated to a specific action at the level of the receivers 5-HT2c renders account of the action activator, contextually to a reduction in the assumption of the food and to a reduction of the subjective hunger, with consequent loss of weight. Sertralina and fluvoxamina interact moreover with the receiving sigma, action that of it suggests the use, in combination to antipsychotic drugs, for the symptoms negatives of the schizophrenia.
Side effects:
Effects collaterals it goes in any case emphasized that the effects are numerous not wroting collaterals in action of this type of drugs, between which those mainly frequent are: loss of appetite (in some cases this induces to use these drugs in the therapy of some alimentary disturbances), nausea, insomnia, tremors, disturbances of the sexuality (reduction of the libido, impotence, anorgasmia). Generally the assumption of these drugs, in particular of the fluoxetina, strongly is adviced against in pregnancy and breast-feeding; or in the event necessary to continue the therapy also in this phase, the choice falls back of norm on the sertralina rather than on other molecules.
The selective inhibitors of the reuptake of serotonin (SSRI), with the same effectiveness of the TCA, but with smaller secondary effects and collaterals of this last. These molecules are unprovided of the blocking activity on the cholinergic, histaminergic receivers H-1 and α1-adrenergic and therefore they do not introduce the effects collaterals of the TCA which had to their affinity for these receivers. The characteristic effects collaterals of this drug class consist in the disturbances of the gastrointestinal sphere (nausea, vomiting, stomachache) and in sexual dysfunctions and are the consequence of the increased serotoninergic activity at the level of the receivers 5-HT2 and 5-HT3.
FLUOXETINA

Description:
Hydrochloride Fluoxetina, commonly said Fluoxetina, is an antidepressant drug used in order to cure the obsessive-compulsive depression, disturbances, abnormal appetite nervous and attacks of panic.
Classification:
Re-uptake selective inhibitor of serotonin (SSRIs)
Therapeutic indications:
The Fluoxetine is indicated for the treatment of depression, obsessive compulsive disorder, bulimia nervosa and the disorder hyperalimentation uncontrolled.
Pharmacokinetics:
The Fluoxetine can be administered during or outside meals because the food does not alter the systemic bioavailability although it may slow down slightly l 'absorption.
Absorption of Fluoxetine after oral administration is rapid and complete. In humans, after a single dose of 40 mg, there are peak plasma Fluoxetine between 15 and 55 ng / ml after 6-8 hours.
Preparations of Fluoxetine capsules, soluble tablets and oral solution are bioequivalent. The Fluoxetine is metabolized in the liver mainly in norfluoxetine and other inactive metabolites subsequently excreted by the kidney. The Fluoxetine is widely distributed in the and is widely linked to plasma proteins. The elimination half-life of fluoxetine is 4-6 days, while that of its active metabolite is 4-16 days (these values can be further prolonged in patients with deficiency of the enzyme P450 2D6). This leads to a significant accumulation of these products in use, chronic active. The presence of liver failure may impede the elimination of Fluoxetine.
In patients with severe renal insufficiency may be a further accumulation of Fluoxetine and its metabolites.
Mechanism of action:
It works by stimulating the proliferation of neuronal precursor cells that are specialized on the step following that of stem, cells that do not yet specialized but now destined to become so. These cells are called "neural progenitors in amplification" (ANP). Fluoxetine also inhibits the uptake of NA and this effect associated with a specific action at receptor level 5HT2C aware of activating, simultaneously with a reduction in recruitment of food and a reduction in subjective hunger, resulting in loss of weight.
Side effects:
Anxiety, nervous tension and insomnia are present in 10-20% of patients. Headache (20%), somnolence (13%), asthenia (9-21%) and tremor (8%) are also found in patients treated with Prozac.
Changes of appetite and weight, significant weight loss, especially in depressed patients under weight may be an undesirable result of treatment. In controlled clinical trials, approximately 9% of patients treated with fluoxetine experienced anorexia. This incidence is approximately 6 times higher than that in the view controls treated with placebo.
Fluoxetine should be administered with caution in patients with a history of seizures.
Platelet function: there have been sporadic reports of altered platelet function in patients taking fluoxetine. Fluoxetine may prolong bleeding time as it reduces the content of serotonin in the granules of platelets, have also been observed petechiae and ecchymosis. The interruption of therapy is followed by a slow recovery of platelet function.
Toxicity:
Studies on reproduction has been made in rats and rabbits at doses 9 and 11 times the maximum daily human dose (80 mg) and did not reveal any detrimental effect to the fetus due to Prozac.
Although studies in animals have not shown any teratogenic or embryotoxic effect selective to the safety of fluoxetine in women during pregnancy has not been established, so the product should not be used during pregnancy unless the potential benefit exceeds the possible risk and still under the direct supervision of the Doctor.
Breast-feeding:
Many drugs are excreted in human milk, including fluoxetine, use particular caution in the administration of fluoxetine in breastfeeding women.
SERTRALINA

Description:
The hydrochloride sertraline is an antidepressant drug prescribed for the treatment of the depression in the patients adults, included the associated depression to anxiety symptoms. This drug is used for the treatment of the symptoms of the depression, obsessive-compulsive disturbance (DOC), of the attacks of panic (associates or not agoraphobia) and of the disturbance from post-traumatic stress.
Classifications:
Re-uptake selective inhibitor of serotonin (SSRIs)
Directions:
The Sertraline is primarily used to treat major depression in adult outpatients as well as the obsessive-compulsive disorder (DOC), panic disorder and social anxiety in adults than in children.
The sertraline is highly effective for the treatment of panic attacks, but the cognitive - behavioral is the best treatment for obsessive-compulsive disorder, combined or not with sertraline administration. For social phobia and post-traumatic disorder Stress (PTSD), the sertraline leads to only modest improvements.
Pharmacokinetics:
In humans, after an oral dose, the peak blood is reached after about 6-8 hours. About 98% of circulating drug is bound to plasma proteins.
It was demonstrated that the pharmacokinetics of sertraline in pediatric patients and adolescents with obsessive-compulsive disorder is similar to that of adults (even in pediatric patients and adolescents the sertraline is metabolized slightly more efficiently). However, taking into account the lower body weight (especially from 6 to 12 years) in order to avoid excessive plasma levels, it is advisable to administer a lower dose to pediatric patients.
The pharmacokinetic profile in adolescents and the elderly do not differ significantly from what is found in adults aged between 18 and 65 years.
The half-life of plasma sertraline is about 26 hours while that of its main metabolite, N-desmetilsertraline, is approximately 62-104 hours. The in vitro activity of N-desmetilsertraline is significantly less than that of sertraline (<20 times) and there is no evidence of its activity in in vivo models of depression.
In the sertraline and N-desmetilsertraline are both extensively metabolised and the resulting metabolites are excreted through the feces and urine in equal amounts. Only a small amount of unchanged sertraline is excreted in the urine. Since the bioavailability of capsules Zoloft increases in the presence of food, it is recommended to administer Zoloft capsules with meals. The food however does not alter the bioavailability of Zoloft tablets. Absorption: The pharmacokinetic profile of sertraline is proportional to the dose of approximately 50 - 200 mg.
After a single oral daily dose of 50 - 200 mg of sertraline for 14 days, the maximum plasma concentrations were reached after 4.5 - 8.4 hours. Based on the percentage recovery in urine and faeces, it can be estimated that absorption after oral administration is at least 70%.
Bioavailability is reduced by the first step. The concomitant intake of food does not significantly influence the bioavailability of sertraline tablets. Distribution: The binding of sertraline with the plasma proteins is about 98%.
The data collected in animal studies indicate that sertraline has a high volume of distribution.
Plasma concentrations of sertraline steady-state (steady state) are thus reached after approximately 1 week and are two times higher than those detected after the initial dose with a single daily dose. Metabolism: Both sertraline that its main metabolite, N-desmetilsertraline, are extensively metabolized in the liver.
The N-desmetilsertraline shows in vitro activity significantly lower than the substance from which it originates.
The metabolite did not show any effect in models of depression in vivo. In experiments in vitro has been shown that the metabolism of sertraline is mediated primarily from enzyme of cytochrome CYP 3A4, with limited involvement of CYP 2D6.
The standard dose of 50 mg sertraline the show only limited effects on the metabolism of other substances mediated by CYP 2D6 and CYP 3A4.
Excretion: The elimination half-life of sertraline average is around 26 hours.
The half-life of N-desmetilsertraline is between 62 and 104 hours, so that plasma concentrations of the metabolite reach the same level of the substance from which it originates. The metabolites of sertraline and N-desmetilsertraline are eliminated in equal fractions in the faeces and urine.
Urine is found only a small percentage (less than 0.2%) of unchanged sertraline. Elderly: The pharmacokinetic profile of sertraline in elderly patients is similar to that in younger patients.
Molecular mechanism:
The sertraline blocks the transport of DA and this effect may explain its properties and activator disinfect detectable in thinking especially in elderly patients, but could lead to an excessive stimulation of the patient, with expressions of anxiety.
Side effects:
Assessment of frequency: very common> 1 / 10, common> 1 / 100, <1 / 10, uncommon> 1 / 1000, <1 / 100, rare> 1 / 10000, <1 / 1000, very rare <1 / 10000; unknown: not estimable from the data available. Erythropoietic system disorders. Uncommon: purpura, altered platelet function, alteration of hemorrhagic diathesis (eg. With epistaxis, gastrointestinal bleeding or haematuria); Rare: leukopenia and thrombocytopenia. Endocrine disorders. Rare: gynecomastia, hyperprolactinemia, galactorrhea, hypothyroidism, syndrome of inappropriate secretion of ADH. Disorders of metabolism and nutrition.
Rare: Hyponatremia: This symptom and disappeared after discontinuation of therapy. Isolated cases may be attributable to the syndrome of inappropriate secretion of ADH. These side effects have occurred predominantly in elderly patients and in patients who used diuretics or other medicinal products. Hypercholesterolemia. Psychiatric disorders. Very common: insomnia, somnolence and anorexia; common yawns, agitation and anxiety; uncommon: euphoria, depressive symptoms, hallucinations, mania and ipomania; rare loss of libido (in women and in humans), nightmares, aggressive reactions psychosis not known: suicidal ideation and suicidal behavior (have been reported cases of suicidal ideation or behavior during treatment with sertraline or shortly after discontinuation of treatment).
Nervous system disorders. Very common: tremor, dizziness and dry mouth; common: headache, motor disorders (including extrapyramidal symptoms such as hyperkinesia, increased muscle tone, bruxism and gait disturbances), paresthesia, hypoaesthesia and increased sweating; uncommon: headache; rare : involuntary muscle contractions, coma, convulsions, acatisia / psychomotor restlessness, signs and symptoms associated with serotonergic syndrome: agitation, confusion, diaphoresis, diarrhea, fever, hypertension, stiffness and tachycardia. In some cases, these symptoms appeared after concomitant use of serotonergic drugs. Diseases of the eye. Common: the reduction of demand; uncommon: mydriasis.
Diseases of the ear and labyrinth. Common: tinnitus. Heart disease. Common: Chest pain and palpitations; uncommon: hypertension, syncope, and tachycardia. Vascular disorders: uncommon: peripheral edema and periorbital edema. Respiratory, thoracic and mediastinal. Rare: bronchospasm. Gastrointestinal disorders: Very common: nausea and diarrhea / soft stools; common: dyspepsia, obstinate constipation, abdominal pain and vomiting; uncommon: pancreatitis and increased appetite. Hepato-biliary diseases. Uncommon: severe hepatic disorders (including hepatitis, jaundice and liver failure) asymptomatic increases in serum transaminases (SGOT and SGPT).
Alterations in the levels of transaminases were reported mainly during the first 9 weeks of treatment and rapidly disappeared after interruption of therapy. Skin and subcutaneous tissue disorders. Common: rash, not common: pruritus, alopecia and erythema multiforme, rare: photosensitivity skin, hives, swelling of Quincke, serious skin desquamation (eg. Stevens-Johnson syndrome and epidermal necrolysis). Diseases of the musculoskeletal system, connective tissue. Uncommon: arthralgia. Renal and urinary disorders.
Uncommon: urinary incontinence; Rare: facial edema, and urinary retention. Pathologies of the reproductive system and breast. Very common: sexual dysfunction (mainly delayed ejaculation in humans); Common: menstrual disorders; rare: priapism. Systemic diseases. Common: asthenia, fatigue and hot flushes; uncommon: indisposition, or increased body weight loss and fever; rare: anaphylactoid reaction, allergic reactions and allergies. Diagnostic tests. Uncommon: abnormal laboratory values. Withdrawal symptoms after discontinuation, the discontinuation of treatment with SSRI / SNRI (especially if sudden) usually leads to withdrawal symptoms. Have been reported dizziness, sensory disturbances (including paraesthesia, electric shock sensation), sleep disturbances (including insomnia and nightmares), agitation or anxiety, nausea and / or vomiting, tremor, confusion, sweating, headache, diarrhea, palpitation, instability 'emotional irritability', visual disturbances.
In general these symptoms are mild to moderate and self-although, in some patients, can be severe and prolonged. Please note that when the efficacy of the treatment is not enough, we must make a gradual riduction dose. In clinical trials for the treatment of social phobias and detected a male sexual dysfunction in 23% of patients (corrected according to placebo). Adverse reactions to drugs depend on the dose administered and are often transient, longer if the treatment continues. Were also reported signs and symptoms characteristic of a serotonergic syndrome as agitation, confusion, diaphoresis, diarrhea, fever, hypertension, stiffness tachycardia and, in some cases associated with concomitant use of serotonergic drugs. More 'than 700 elderly patients (with more than 65 years)
participated in a clinical study that proves the efficacy of sertraline in these patients. The type and frequency of side effects in elderly patients were similar to those observed in most patients' young people.
Toxicity:
Were not shown teratogenic or embryotoxic effects in any of the doses used. Dose corresponding to about 2,5-10 times the maximum human dose, however, sertraline caused maternal toxicity in a position to delay the succession process of fetal ossification. There are no adequate studies in pregnant women.
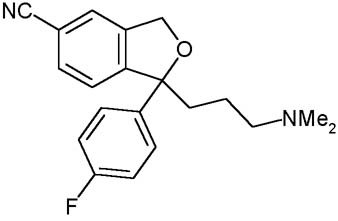
CITALOPRAM
Description:
The citalopram it is a molecule of the family of antidepressants not tricyclic SSRI. The citalopram it is the more recent molecule and, to said of numerous studies, most selective and, consequently, that which it introduces less effects collaterals. It comes widely employed, date its elevated tolerability, in the light depressive syndromes and the disturbance of panic to dosages comprised between the 20 and 40 mg. Currently it is the most powerful inhibitor of the receivers of serotonin in commerce, but not acting directly on they, how much on the king-uptake of the neurotransmitter, than remaining inhibited it more over a long time span maintains the serotonin at the level of the synaptic fissure.
The citalopram is an antidepressant belonging to the class of selective inhibitors of serotonin reuptake (SSRIs).
The pharmacological action of citalopram is expressed through inhibition of serotonin re-uptake and a down-regulation of postsynaptic serotonin receptors after prolonged administration. The citalopram shows little interaction with the alpha-adrenergic receptors, and anticholinergic histaminic. The drug is rapidly absorbed after oral administration with the attainment of maximum plasma levels within 2-4 hours after administration and the bioavailability is almost complete. Has some active metabolites that, while the weakest of the compound of origin and with very low plasma levels, retain selective serotonergic action. The plasma half-life is approximately 36 hours and the binding protein is less than 80%. The pharmacokinetics of citalopram is a linear, ie, the plasma level increases in proportion to increasing dose and is not influenced by age. The achievement of steady state occurs in about 1 weeks. The citalopram is indicated for the treatment of depression and obsessive-compulsive disorder.
Further information regarding the other disorders linked to serotonergic spectrum, such as food conduct disorders, disorders of the behavior of aggressive or impulsive type, other anxiety disorders (eg. This disorder from panic attacks), headaches and chronic pain, etc.. The dosage range of citalopram in the treatment of depression is usually between 20 and 40 mg / day. In obsessive-compulsive disorder dosing tends to take place in the higher range, while in disorder from panic attack the initial prescription would be around 10 mg / day, with increments at the discretion of the physician, based on clinical response.
The side effects of the drug, like all SSRIs, are linked to increasing serotonergic tone in acute outweigh the side effects of gastrointestinal type (in 15-30% of cases may occur transiently nausea, iporessia rarely gastralgie, vomiting, diarrhea or soft stools) and side effects related to the action of serotonin on the CNS (in 10-15% of cases may occur transiently irritability, insomnia, sedation, tremors, etc.).. With lower frequency phenomena are related to the action of serotonin in peripheral vascular, such as headache and sweating. To treat chronic disorders tend to occur of sexuality, especially the delay in reaching orgasm (sometimes used in treating male Premature Ejaculation) and reduced libido.
The use of citalopram is contraindicated in the course of contemporary drug taking monoamino oxidase inhibitors (MAOIs), as were observed serious reactions, sometimes fatal. The citalopram, like all SSRIs, although not showing the reactions of direct cardiotoxic tricyclic antidepressants should be used initially reduced dosage and careful clinical monitoring for the risk of transient response vasoconstrictive, individual-employee, in the heart attack acute myocardial infarction. The drug has shown inhibition of the CYP450 system, less than other SSRIs, which manifests itself mainly dependent type of isoenzymes 2D6 and 3A4. Plasma levels of citalopram slightly higher were observed during treatment with some phenothiazines. Plasma levels of lithium should be monitored when it is associated with the citalopram or other SSRIs. Further caution should be used in association between SSRIs and other drugs acting on the serotonergic system (eg., Serotonin precursors like 5-idrossitriptofano, other serotonin-type antidepressants, lithium salts, etc..) As may, in susceptible persons, manifest a serotonergic syndrome.
Classification:
Re-uptake selective inhibitor of serotonin (SSRIs)
Directions:
Endogenous depressive syndromes and disorders' s crisis of anxiety with panic, with or without agoraphobia.
Citalopram is a bicyclic ftalenic derivative with antidepressant effect.
Biochemical and behavioral studies have shown that pharmacodynamic effect of citalopram is closely related to uptake potent inhibition of 5-HT (serotonin).
The citalopram has no effect on uptake of NA (noradrenaline) and is therefore the most selective inhibitor of serotonin dell'uptake so far described.
The citalopram has no influence on uptake of DA (dopamine) or GABA (gamma-aminobutyric, acid). Moreover, it citalopram nor its metabolites have propriety antidopaminergic, antiserotoninergic, or anticholinergic, antihistaminergic and not inhibit the MAO (monoamino oxidase).
Citalopram does not bind to benzodiazepine receptors, GABA, or opioid.
After prolonged treatment, the uptake inhibitory effect of 5-HT is unchanged, in addition the citalopram does not induce changes in the density of neuroreceptor as with most of the tricyclic antidepressants and the newer atypical antidepressants.
There are no effects on muscarinic cholinergic receptors on histamine receptors and alpha-adrenoreceptor, resulting in no occurrence of side effects related to inhibition of these receptors: dry mouth, sedation, orthostatic hypotension, which are present after treatment with several antidepressant drugs. Citalopram is unique because of its extreme selectivity and blocking to uptake for the absence of agonist or antagonist activity on the receptors.
Pharmacokinetics:
Citalopram is rapidly absorbed after oral administration. The maximum plasma levels are reached within 2-4 hours after administration.
The apparent volume of distribution is approximately 14l1kg (range 12-16l1kg). The legarne to plasma proteins is less than 80%.
The bioavailability of citalopram after oral administration is practically complete.
It has been demonstrated a linear relationship between plasma concentrations at steady state and the dose administered, with average concentrations of 250 nM for a daily dose of 40 mg.
The biological half-life is approximately one and a half days and in most patients the steady state is reached within the first week of therapy.
In most patients the levels of steady-state are included in the range 100-400 nM for a daily dose of 40 mg.
In elderly patients because of low speed metabolism, were found a half-life longer and clearance decreased.
Like other psychotropic drugs, is distributed in the citalopram and the highest concentrations of drug metabolites and demethylation can be found in the lungs, liver, kidneys, lower concentrations in the spleen, heart and brain. The drug and its metabolites pass the placenta and the fetus are distributed in a similar way to what is seen in the mother.
A very small amount of citalopram and its metabolites are secreted in breast milk. The citalopram is metabolized to dimethylcitalopram, di-dimethylcitalopram, citalopram N-oxide and, for deamination, a derivative of propionic deaminate.
While the derivative of propionic acid are inactive dimethylcitalopram, di-dimethylcitalopram and citalopram N-oxide, are also selective inhibitors of serotonin to uptake, although the weakest of the compound of origin.
In patients, citalopram is not metabolized the predominant compound in plasma.
The ratio of concentration citalopram / dimethylcitalopram in plasma at steady state, an average of 3.4 after 15 hours and 2 after 24 hours after administration.
In the plasma di-dimethylcitalopram and citalopram N-oxide are generally very low.
No assessment was carried out between plasma concentration and effect, no side effects appear to be related to plasma concentrations of the drug. The systemic plasma clearance is approximately 0.4 l / min. The excretion occurs in urine and faeces.
The conversion factor from nM to ng / ml (referring to the base) is 0.32 for citalopram and 0.31 for dimethylcitalopram.
Molecular mechanism:
The citalopram is the most selective of the SSRIs, does not interact with other carriers, has only a weak affinity for the regard to histamine H1 receptors, does not inhibit the activity of CYP 450 and therefore does not give rise to dangerous interactions with other drugs.
Side effects:
Citalopram is the molecule the more recent and more selective, and one that has less side effects. It is widely used because of its high tolerability in mild depressive syndromes and panic disorder at doses between 20 and 40mg.
Toxicity:
The safety of citalopram during pregnancy has not been established. Although studies conducted on experimental animals have not shown signs of potential teratogenicity, it effects on reproduction or perinatal conditions, as the citalopram and its metabolites pass the placenta and because a very small amount is found in breast milk, we do not use during pregnancy and lactation.
ESCITALOPRAM

Description:
Escitalopram is a used active principle in antidepressant drugs, as it has the property to inhibit the reuptake, in the inter-synaptic neuronal spaces, of the serotonin, a neurotransmitter in charge of the regulation of the tone the humor in individual. Assuming such molecule a greater amount of serotonin is had, on hand of the brain, consequently will be had a greater stabilization of humor in subjects typically depressed or tending to the depression. The assumption of this drug, so that the relative stabilization happens of humor, will have to be rather extended in the time, for to have turned out long-lasting. The first result will beginning from have 2 or 3 the week of assumption; during 1e 2 the week probable effects collaterals are:
increase of weight, nausea, dryness of makes us and of the language, cephalalgy and light insomnia.
The escitalopram (Cipralex, ENTACT) is an active ingredient used in antidepressant drugs, as has the property of inhibiting the re-uptake in inter-neuronal spaces synaptic, serotonin, a neurotransmitter responsible for setting the mood of the individual tone . Assuming that the molecule is a larger amount of serotonin available to the brain, therefore you will have a greater stabilizing the mood in subjects typically depressed or prone to depression.
Taking this drug, that occurs on the stabilization of mood, must be quite extended in time, to have lasting results.
The active enantiomer of citalopram, escitalopram over the four times to be more selective of the racemic mixture, minimally inhibits the activity of CYP 450 and therefore does not give rise to dangerous interactions with other drugs. In vivo studies show that escitalopram possesses a pharmacological equivalent to or greater than that of a double dose of citalopram. All studies conducted in animals in vivo experiments confirm that the effects of citalopram are due an enantiomer S, while the R enantiomer, would not only be devoid of pharmacological activity, but the effects attenuate of enantiomer active. Recent kinetic studies of the link radioreceptorial have allowed to assume the specific mechanism of interaction of the two enantiomers of citalopram with serotonin transporter responsible for the characteristic effect of the racemic mixture compared to only the active enantiomer.
Due to its molecular structure the escitalopram is able to bind to both the primary site of high affinity, thereby blocking the transport of serotonin, is the site of modulation allosteric resulting in a conformational change of the protein that makes a more stable binding of escitalopram to primary site, thus leading to a complete blockade of the transporter.
Classifications:
Re-uptake selective inhibitor of serotonin (SSRIs)
Directions:
Treatment of major depressive episodes, treatment of eating disorders from panic attacks with or without agoraphobia.
Pharmacokinetics:
Absorption is almost complete and independent from the intake of food (the average time to maximum concentration (Tmax average) is 4 hours after multiple doses.
It is expected that the absolute bioavailability of escitalopram is approximately 80%, as for the racemic compound citalopram. The apparent volume of distribution (Vd, β / F) after oral administration is approximately 12-26 L / kg.
The plasma protein binding is less than 80% for escitalopram and its main metabolites. Regarding the metabolism Escitalopram is metabolised in the liver and demethylation metabolites in di-dimethylation.
Both are pharmacologically active.
Both the origin of drug metabolites are partly excreted as glucuronide.
Following multiple doses of the average concentrations of demethylation and di-dimethyl metabolites are 28.31% and <5%, respectively, the concentration of escitalopram.
The biotransformation of escitalopram in demethylation metabolite is mediated primarily by CYP2C19.
It is possible contribution of the enzymes CYP3A4 and CYP2D6.
Elimination: the elimination half-life (t ½ β) after multiple doses is approximately 30 hours and the oral plasma clearance is approximately 0.6 L / min.
The major metabolites have a significantly longer half-life. It is expected that escitalopram and its main metabolites are eliminated by both routes, hepatic (metabolic) and renal, with most of the dose excreted as metabolites in urine. The pharmacokinetics is linear type.
Plasma levels at steady state is reached in about 1 week. The average concentrations of 50 nmol / L (range 20 to 125 nmol / L) at steady state are achieved with a daily dose of 10 mg. Elderly patients (> 65 years) escitalopram seems eliminated more slowly in the elderly compared with younger patients.
The systemic exposure (AUC) in the elderly is approximately 50% higher compared to young healthy volunteers. The liver function is reduced in patients with mild hepatic dysfunction or moderate, the half-life of escitalopram is about two times longer and exposure of about 60% higher than in patients with normal liver function. In the racemic compound citalopram was observed a longer half-life and a minor increase of the in patients with impaired renal function.
Plasma concentrations of metabolites were not studied, but could be high. Polymorphism has been observed that the slow metabolizers, compared with CYP2C19, have a plasma concentration of escitalopram two times higher than the fast metabolizers.
No significant change in exposure was observed in slow metabolizers than in CYP2D6.
Mechanism of action:
Due to its molecular structure the escitalopram is able to bind to both the primary site of high affinity, thereby blocking the transport of serotonin, is the site of modulation allosteric resulting in a conformational change of the protein that makes a more stable binding of escitalopram to primary site, thus leading to a complete blockade of the transporter. The R enantiomer is not able to bind to the primary site, it is a weak inhibitor of the serotonin transporter, but binds to the site allosteric in such a way as to destabilize the binding site S of enantiomer primary and interfering with inhibitory activity of escitalopram. Therefore, the administration of the racemic mixture citalopram does not produce a complete blockade of the transporter, since the presence of enantiomer R partially reduces the power of enantiomer S. This particular effect of enantiomer inactive account would make the profile of the bell curve dose-response relationship of citalopram, ie a reduction of the higher doses, and the fact that, in several preclinical studies, escitalopram demonstrated higher activity that of the corresponding double dose of citalopram.
Side effects:
The escitalopram is the evolution of citalopram and, consequently, more selective, with fewer side effects. The effects are the same as the citalopram and found to have similar frequency, the most common are nausea, diarrhea, dry mouth, insomnia or drowsiness, dizziness, hyperhidrosis, sexual dysfunction, especially Premature disorders (9%), but also decrease libido (4%) and impotence (3%).
Toxicity:
In reproductive toxicity studies in rats performed with escitalopram were observed embryo-foetus toxic but no increase in the incidence of malformations. The risk in humans is unknown. It should not be used during pregnancy unless absolutely necessary and only after a careful assessment of risk / benefit.
FLUVOXAMINA
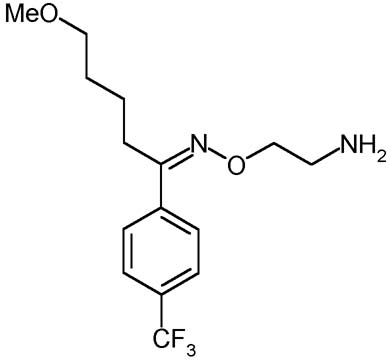
Description:
The fluvoxamina is a selective inhibitor of the reuptake of serotonin (SSRI). It is used mostly for the treatment of the depression and obsessive-compulsive disturbance (DOC). Although that its effects are partially similar to those of the other SSRI, the fluvoxamina has different farmacologic effects. For this reason it can be used in order to create a benefit in the not usual patients who have tried effects limiting collaterals. On the whole the Fluvoxamina seems to cause to smaller effects collaterals regarding the other SSRI, above all in relation to the loss of sexual desire; moreover this molecule associates an antidepressant action an effect anxiolytic thus becoming useful in the forms of expectant depression. Still the fluvoxamina is recognized to be a drug of great effectiveness in the depressions in course of psychosis, and the manifestations of deficit of the control of the impulses. The fluvoxamina does not have to be administer in combination with inhibitors of monoammino oxidase (IMAO), thus as it is adviced against the association with tricyclic antidepressants, of which it increases the levels put molding on to us. It moreover is an inhibitor of the CYP450 1A2 that metabolize the tricyclic, caffein, teofillina, clozapine, tacrina, antidepressants, paracetamol and the phenacetin.
Classifications:
Re-uptake selective inhibitor of serotonin (SSRIs)
Directions:
Molecule linking antidepressants to an anxiolytic effect. It is therefore particularly useful in forms of anxious depression (300 mg.). Fluvoxamine may also be considered a drug of considerable effectiveness in treating obsessive-compulsive disorder (250-300 mg.) And in depressions in the course of psychosis, as well as in the manifestations of deficiency of the control pulse.
Pharmacokinetics:
The fluvoxamine is rapidly and completely absorbed after oral administration. In vitro, the plasma protein binding is equal to about 77%. The mean plasma half-life is approximately 15 hours after single administration and slightly longer (17 - 22 hours) after repeated dosing. Constant blood levels are reached within the first two weeks of treatment at doses constant. The drug is metabolized by the liver primarily by oxidative demethylation with formation of metabolites eliminated by the kidneys. The two main metabolites were devoid of pharmacological activity. The pharmacokinetic profile in elderly patients (> 60 years) is similar to that observed in young subjects (20-35 years).
Mechanism of action:
The fluvoxamine, among the SSRIs, is the molecule with characteristics more sedative, and is a potent inhibitor of 1A2 and 3A4 isoforms of CYP450 and thus may lead to interactions with other drugs that follow the same metabolic pathway. Fluoxetine also inhibits the uptake of NA and this effect associated with a specific action at receptor level 5HT2C aware of activating, simultaneously with a reduction in recruitment of food and a reduction in subjective hunger resulting in weight loss.
Side effects:
The main side effects of fluvoxamine are nausea, sleepiness, insomnia, agitation, nervousness, dizziness, tremors, anorexia, dyspepsia, constipation, diarrhea, xerostomia, sweating, palpitations, headache, asthenia.
Rischio ictus negli anziani in trattamento con SSRI
Rischio ictus negli anziani in trattamento con SSRI Nei pazienti anziani trattati con antidepressivi, rispetto a coloro che non li assumono, soltanto gli inibitori selettivi della ricaptazione della serotonina (Ssri) appaiono associati a un aumento del rischio di ictus ischemico, soprattutto sul breve termine. Questo il verdetto di uno studio caso-controllo di popolazione condotto da Gianluca Trifirò e collaboratori del Dipartimento di Informatica medica del Centro medico Erasmus di Rotterdam. La ricerca si è basata su dati, relativi al periodo 1996-2005 e ricavati dall'Integrated Primary Care Information database, di pazienti di età >/= 65 anni colpiti da un primo ictus ischemico posti a confronto con soggetti paragonabili per anno di nascita, sesso e data indice. L'esposizione ai farmaci antidepressivi è stata suddivisa nelle categorie "in corso", "trascorsa", "nessun impiego" e la terapia in base al tipo di farmaco (Ssri, triciclici e altri antidepressivi), alla dose e alla durata. Il confronto tra il rischio di ictus ischemico di utilizzatori e non utilizzatori di antidepressivi è stato eseguito mediante regressione logistica condizionale. In totale sono stati identificati 996 casi di ictus ischemico incidente. Per quanto riguarda gli Ssri, nei pazienti anziani la terapia in corso è risultata associata a un significativo incremento del rischio di ictus rispetto al non-uso (odds ratio 1,55), soprattutto quando i farmaci venivano assunti da meno di sei mesi. Nessuna associazione con il rischio ictus, invece, è stata registrata per la terapia in corso con triciclici o altri antidepressivi.
J Clin Psychopharmacol, 2010; 30(3):252-8