INTRODUCTION
Thromboembolic disease is the third most common vascular disease ( after CHD and stroke) in the developed world, and is caused by an excessive stimulation of coagulation.
That is due to the presence of one or more of the three class of factors identified in XIX century by Virchow :
1) Alterations in blood’s composition
2) Local alterations in the physiological blood flux
3) Alterations and/or lesions of the endothelium
Many patients, with several different clinical conditions, requires therefore an anticoagulant treatment.
Patients affected by
Atrial fibrillation, hypercoagulable conditions, mechanic heart valves carriers, dialysed patients and those undergoing endovascular procedures or cardiothoracic/vascular surgery, are therefore treated with thrombin indirect inhibitors (heparin –LMWH) or vit K antagonists (warfarin ).
Those agents are clinically effective, although presenting characteristics that prevent them from providing optimal anticoagulation and convenience.
Actually, heparin limitations are:
• Need for antithrombin as cofactor present in patient’s plasma to achieve its therapeutical effectiveness
• only injectable administration drug- not suitable for long term anticoagulation therapy
• A tendency to bind various plasma proteins, creating an unpredictable dose-dependent anticoagulant effect. Need for routine dose –adjustement and coagulation monitoring.
• Inability of the heparin – antithrombin to bind and inhibit thrombin if bound to fibrine ( further thrombus growth if still present at the administration time.
• Heparin- induced thrombocytopenia (HIT ) as complication.
While Warfarin ones are:
• Narrow therapeutic window
• Slow onset of action and difficult compliance
• Interaction of Warfarin with several drugs and some foods, (intestinal flora variations), makes necessary a monitoring of their international normalized ratio ( in range if between 2.0 and 3 if patients affected by VTE or AF; 2.5-3.5 if patients with mechanic heart valves)
• Possible side effect as skin necrosys and bleeding, especially intracranial bleeding
• Teratogenic
Hence, new improved oral anticoagulants are being investigated to optimize the balance between efficacy and safety.
One of the results of such an effort, was the development of new direct thrombin inhibitors.
DIRECT THROMBIN INHIBITORS

Figure 1: coagulation cascade

Figure 2: Thrombin (or Factor IIa)
Thrombin is a key serine protease involved in the coagulation cascade processes.
Its precursor is Prothrombin, codificated on chromosome 11 (11p11-q12); Prothrombin is proteolitically cleaved to form thrombin by factor Xa , activated either by intrinsic and by extrinsic pathway, and by factor V .
The molecular weight of prothrombin is approximately 72,000 Da .
The catalytic domain is released from prothrombin fragment (3) to create the active enzyme thrombin, which has a molecular weight of 36,000 Da.
Its activity is inhibited by endogenous circulating anticoagulants, such as Antithrombin (AT), Heparin cofactor II (HCII) and finally binding to the cofactor Trombomodulin ™, it induces protein C activation (8) (Griffin 1995 the thrombin paradox )
Thrombin’s main function is to convert soluble fibrinogen to insoluble fibrin .
It can also stimulate platelet activation.
Furthermore, thrombin also activates factors V, VIII, XI (enhancing protrombin to thrombin conversion) and XIII (clot stabilization).
Finally, thrombin can induce several cellular effects also by activating a family of G-protein coupled protease activated receptors (PAR 1,2,3 and 4)
PAR1 is expressed by either physiological and pathological endothelium, suggesting a relevant thrombin’s role on it.

Figure 3 - Interactions of six different anticoagulant with thrombin (5)
Thrombin can be inhibited directly or indirectly by the binding of thrombin inhibiting drugs to one or two of its three domains: the active site and exosites 1 and 2 (see figure 3).
Exosite 1 is the fibrin binding site of thrombin and exosite 2 serves as the heparin-binding domain (5)
Traditional anticoagulants such as unfractionated heparin (UFH) and low-molecular weight heparin (LMWH) inhibit free thrombin in an indirect manner by binding simultaneously to antithrombin and exosite 2, thereby forming a heparin-thrombin antithrombin complex (Figure 3).
Direct thrombin inhibitors (DTIs) instead, bind directly to thrombin and do not require a cofactor such as antithrombin to exert their effect.
This direct bound to the active site distinguish them from indirect thrombin inhibitors, such as Heparin and LMWH.
On the other hand, no antidotes to contrast their action in case of toxicity are known.
To quantify direct thrombin inhibitors, an universal effective test is Ecarin clotting time (Nowak G.; 2004 )
Figure 4 – Hirudo medicinalis
The forerunner of the class can be considered hirudin, originally isolated in the saliva of leeches (Hirudo medicinalis) (figure 4).
Are distinguished bivalent DTI (binding both to the active catalytic site of thrombin, as well as a site reconnaissance of the substrate) and monovalent ones if they only bind the active site of thrombin.
Among the first class, are included Hirudin, Bivalirudin, Lepirudin and Desirudin.
Among the latter however are counted Argatroban, Melagatran and Dabigatran.
DTIs can inhibit both soluble thrombin and fibrin-bound thrombin.
Moreover, Dabigatran is currently the only DTI orally administrable.

Figure 5: Hirudin in complex with Thrombin
HIRUDIN is an irreversible direct thrombin inhibitor.
It is a peptide of 65 amino acids, organised into a compact N-terminal domain containing three disulfide bonds and a C-terminal domain that is completely disordered when the protein is un-complexed in solution.
Amino acid residues 1-3 form a parallel beta- strand with residues 214-217 of thrombin, the nitrogen atom of residue 1 making a hydrogen bond with the Ser-195 O gamma atom of the catalytic site.
The C-terminal domain makes numerous electrostatic interactions with an anion-binding exosite of thrombin, while the last five residues are in a helical loop that forms many hydrophobic contacts.
Natural hirudin contains a mixture of various isoforms of the protein.
By its recombination, Lepirudin and Desirudin were obtained.
Both recombinant hirudins (r-hirudins) are bivalent direct thrombin inhibitors that bind simultaneously to the active site and exosite 1 domain on thrombin, an interaction that increases their specificity for thrombin. They also have the highest affinity for thrombin as they rapidly form essentially irreversible, 1:1 stoichiometric complexes.
Compared with hirudin their affinity is 10 times weaker for thrombin, however, they are still considered the most potent of all the thrombin inhibitors.
The plasma half-life of the r-hirudins after intravenous (i.v.) injection and subcutaneous (s.c.) administration is 60 and 120 min, respectively.
LEPIRUDIN is licensed for the treatment of thrombosis complicating HIT .
Because of its prevalent renal excretion, a dose adjustment is needed if infused in patients with renal impairment.
Limitations to its use: narrow therapeutic window, increased bleeding events and the absence of a specific antidote to reverse its effect in case of major life threating bleeding events.
Moreover 40% of patients develops anti-Lepirudin Ab during treatment. This lead to Icox development and delayed renal excretion.
In a few cases, anaphilaxis could appear in case of re-exposition to hirudin.
DESIRUDIN The unique DTI approved by FDA for postoperative prevention of VTE in patients undergoing elective hip replacement (subcutaneous administration)
Maximum plasma concentration after 1-3 h; terminal half life of 2 h; mainly renal excreted (80-90%)
Dose adjustement needed for patients with serious renal impairment ( CLcr <30 ml/min) Not necessary if moderate renal impairment (30-60)
BIVALIRUDIN
Hirudine bivalent analogue.
Bivalirudin is a bivalent 65-amino acid single-chain polypeptide linked by
three disulfide chain. Its molecular mass is 2.180 kDa.
Its 20-amino acid structure, combines a carboxy-terminal region that recognises thrombin’s fibrin(ogen)-binding site, and an amino-terminal tetrapeptide that inhibits the active site of thrombin, connected by a tetraglycine spacer.
Because of that, bivalirudin has a bivalent and reversible direct inhibitor effect on thrombin.
This drug is given intravenously, has an immediate onset of action; therapeutic activated clotting time reached in 5 minutes since administration.
Half life of 25 minutes. Clearence of Bivalirudin is mediated by proteolytic cleavage and hepatic metabolism. By the way, 20% of the dose is renally eliminated, requiring a dose adjustments in patients with moderate renal impairment.
Controindicated in patients with severe renal impairment.
FDA approved bivalirudin for the following clinical conditions:
• Alternative anticoagulant therapy to heparin in patients undergoing PTCAs (2000) (32)
• Patients undergoing to urgent PCI procendures (2005) (32)
• Alternative to heparin in HIT patients undergoing PCI
ARGATROBAN
It’s a small univalent molecule (527 Da), directly and reversibly binding the thrombin’s active site.
Intravenous administration is followed by rapid clearance; half life is about 45’ (39’ to 50’).
Argatroban is hepatically metabolized and the main excretion way is biliary system.
Controindicated in case of serious hepatic impairment.
This drug is monitored in its anticoagulating activity by dosing aPTT.
Possibly, it could interfere with warfarin in case of combined therapy alterating INR.
In those cases, therapy can be discontinuated if INR rise over 4
Argatroban’s indication are
• the treatment patients affected by HIT , in presence or absence of thrombosis.
• For patients undergoing to PCI
Notice that the choice between argatroban and lepirudin for HIT’s treatment, is guided by the functional state of liver and kidney.
If a renal failure is detected, argatroban is the best choice.
On the other hand, Lepirudin might be preferred in case of hepatic impairment.
| Pharmacological and clinical proprieties of the parenteral direct thrombin inihibitors |
| Lepirudin | Desirudin | Bivalirudin | Argatroban |
| Indication | Prophylaxis or treatment of thrombosis complicating HIT | DVT prevention after THR | Patients with UA undergoing PTCA;
PCI with provisional use of GPI;
patients with/at risk of HIT/HITTS undergoing PCI | Prophylaxis or treatment of
thrombosis complicating HIT;
patients with/at risk of HIT undergoing PCI |
| Thrombin affinity (Ki) | Highest (Ki = 0.23 pM) | Highest (Ki = 0.26 pM) | Intermediate (Ki = 1.9 nM) | Lowest (Ki = 5–38 nM) |
| Route of Administration | Intravenous, subcutaneous | Intravenous, subcutaneous | Intravenous | Intravenous |
| Dosing | Bolus of 0.4 mg kg-1 followed by 0.15 mg kg-1 h -1
Monitor with aPTT for target ratio 1.5–2.5 | s.c. injection 15 mg twice daily
No monitoring necessary | Bolus of 0.75 mg kg-1
followed by 1.75 mg kg-1 h -1 ¥ 4 h
Monitor with ACT after 5 min (365 100 s) | No bolus followed by 2 mg kg-1 h -1
Monitor with aPTT for target ratio 1.5–3.0 |
| Plasma half-life | ~80’ | Intravenous, 60 min;
s.c., 120 min | ~25’ | ~ 45’ |
| Metabolism | Kidney | Kidney | Proteolysis; liver | Liver |
| Clearence | Kidney | Kidney | 20% excreted via kidney | Liver |
| Dose reduction | CLCR 15–60 ml min-1: 15–50%
CLCR <15 ml min-1: contraindicated | *CLCR <60 ml min-1: dose adjustment necessary | CLCR 15–60 ml min-1 : 15–50%
CLCR <15 ml min-1 : contraindicated | Hepatic failure: 0.5 mg kg-1 min-1
Severe HF: contraindicated |
Table 1: comparison between different ev/sc DTIs
ORAL DIRECT THROMBIN INHIBITORS
Oral IIa inhibitors represent a new era of anticoagulation for the prevention and treatment of venous and selected arterial thromboembolisms.
XIMELAGATRAN is the oral double prodrug of melagatran and was the first oral direct thrombin inhibitor developed(Astrazeneca; Gustafsson et al 2004).
Studies demonstrated it to be as effective as the traditional anticoagulants in the prevention and treatment of venous and arterial thromboses and secondary prevention of cardiovascular events post MI.
After its approval in Europe however, Ximelagatran was removed from the market approximately 20 months later.
Actually, further studies showed that therapy greater than 35 days was associated with a risk of hepatotoxicity (8).
Like ximelagatran, DABIGATRAN ETEXILATE is an orally active double prodrug that is rapidly converted to
dabigatran, a low-molecular weight molecule that acts as a specific, potent and reversible direct thrombin inhibitor.
It is currently the most studied and promising of the oral direct thrombin inhibitors. Key clinical advantages of this drug include a rapid onset of action, lack of interaction with cytochrome P450 enzymes or with other food and drugs, excellent safety profile, lack of need for routine monitoring, broad therapeutic window and a fixed-dose administration.
DABIGATRAN ETEXILATE
DESCRIPTION
Dabigatran is a synthetic , reversible DTI with high affinity and specificity for its target binding both free and clot-bound thrombin , and offers a favourable pharmacokinetic profile.
Furthermore, it is the only DTI administered orally nowourdays approved by FDA.
CHEMICAL STRUCTURE
Dabigatran was synthetizedas a derivate of the peptide-like benzamine-based thrombin inhibitor AA-[alpha] alpha]-naphthylsulphonylglycyl-4-
amidinophenylalanine piperidine ([alpha]-NAPAP).
The chemical name for dabigatran etexilate mesylate, a direct thrombin inhibitor, is b-Alanine, N-[[2-[[[4-[[[(hexyloxy)carbonyl]amino]iminomethyl] phenyl]amino]methyl]-1-methyl-1H-benzimidazol-5-yl]carbonyl]-N-2-pyridinyl-,ethyl ester, methanesulfonate.
The empirical formula is C34H41N7O5 • CH4O3S and the molecular weight is 723.86 (mesylate salt), 627.75 (free base).
The structural formula is:
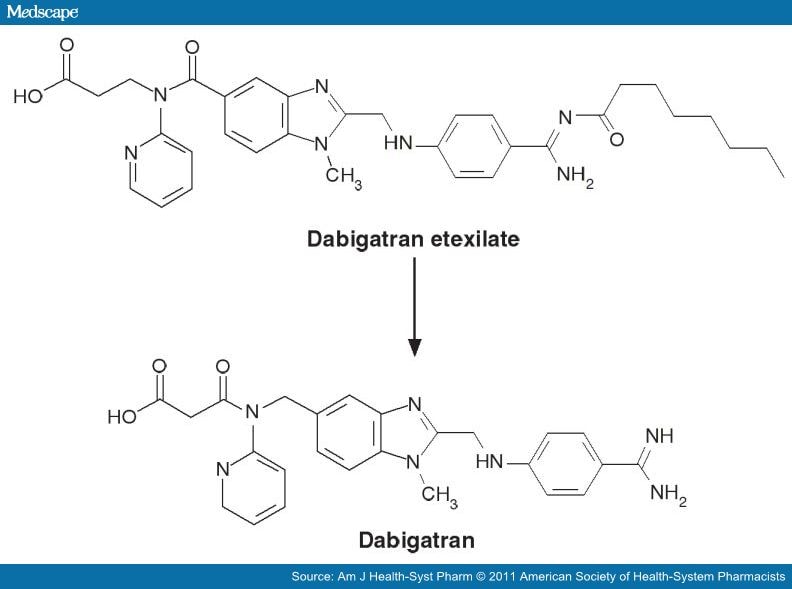
Figure 6: Dabigatran Etexilate and Dabigatran (5)
INDICATIONS
Dabigatran is approved to reduce the risk of stroke and systemic embolism in patients with non-valvular atrial fibrillation (1) (2)
with one or more of the following risk factors;
*previous stroke, TIA or systemic embolism
*Left ventricular ejection fraction<40%
*Symptomatic heart failure of NYHA class 2 or above
*Age>75 years old
*Age ≥ 65 years old associated with one of the following conditions:
diabetes mellitus ; coronary arthery disease or hypertension
Dabigatran has also been approved as an alternative to enoxaparin for prevention of venous and arterial thombo-embolic disorders after orthopaedic surgery (4) (2)
DOSING
According to RE-LY stage III clinical trial, (5)
150 mg dose twice daily was associated with a similar incidence of major
bleeding side effect as warfarin, but patients had better outcome with a
reduction of the incidence of systemic thrombo-embolism or stroke.
110 mg dose twice daily, had a better safety profile compared with warfarin
(major bleeding reduction: 2,71% per year(110 mg dabigatran) vs 3,36%
per year (warfarin))and a similar outcome about incidence of systemic
thrombo-embolism or stroke.
A pharmacokinetic simulation (Clement et al. , 2012) (6), showed that twice daily dosing of dabigatran in AF patients, reduce the plasmatic variations on drug’s concentration (6), being therefore more effective in thromboembolism prevention. (5)
DOSE ADJUSTEMENT
For patients with creatinine clearance (CrCl) > 30 mL/min, the recommended dose of Dabigatran is 150 mg given orally twice daily.
For patients with severe renal impairment, follow the recommended doses:
For patients with CrCl 15-30 mL/min, the recommended dose is 75 mg orally twice daily (8) (“FDA”:http://www.fda.gov/Drugs/DrugSafety/ucm326580.htm) .
Dosing recommendations for patients with a CrCl <15 mL/min or on dialysis cannot be provided. ( “FDA”:http://www.fda.gov/Drugs/DrugSafety/ucm326580.htm )
In Europe, therapy with dabigatran is contraindicated if creatinine clearance is <to 30 ml/min. (8) (2)
For patients who needs to discontinue dabigatran temporarily for invasive procedures, are recommended to stop drug for 1-2 days before intervention (6)
A lower dose can be considered in eldery patients (2)
USE OF DABIGATRAN SHOULD BE AVOIDED IN THE FOLLOWING CONDITIONS:
*Patients with mechanical prosthetic heart valves
*Patients with atrial fibrillation caused by heart valve problems
*Patients with renal severe impairment (creatinine clearence <15 ml/min)
*Patients with active significative bleeding, haemostasis impairment
*Hepatic severe impairment.
*Concomitant treatment with ketoconazole, cyclosporine, itraconazole or
tacrolimus. (1)
*Poor patient’s compliance (5)
Caution should be used when prescribing dabigatran to obese patients, children or pregnant women because of the lack of study and data about the drug’s effect on those particular populations. (10)
POTENTIAL FUTURE DABIGATRAN’S INDICATIONS
Several new indications as oral anti coagulant such as treatment of acute trombo-embolism (4) and HIT’s therapy are being enquired
Secondary prevention of acute coronary infarction in patients with acute coronary syndrome (8) (Oldgreen et al, 2011 )
Reduction of severity of sepsis and infections by Staphylococcus aureus, inhibiting the staphylocoagulase with Dabigatran (9)
Treatment of pathological conditions such as cancer, fibrosis and atherosclerosis, in which it seems to be important the role played by thrombin and PAR1 (8)
CLINICAL PHARMACOLOGY
Mechanism of Action
Dabigatran directly inhibits human thrombin in a concentrative-dependent and competitive way, with a Ki of4.5nM.
This inhibition is rapid, reversible and higly selective for thrombin (8)
Dabigatran bind thrombin directly on the active site (see figure 2 and 3), so unlike LMWH it can inhibit both free and bound thrombin, preventing thrombus development.
Dabigatran, by inactivating thrombin on platelets surface, also prevent thrombin-induced platelets aggregation (8).
It has no effect on platelets aggregation induced by arachidonic acid, collagen and ADP.
PHARMACODYNAMICS
At recommended therapeutic doses, dabigatran etexilate prolongs the coagulation markers such as aPTT, ECT, and TT. INR is relatively insensitive to the exposure to dabigatran and cannot be interpreted the same way as used for warfarin monitoring.
The aPTT test provides an approximation of DABIGATRAN’s anticoagulant effect. The average time course for effects on aPTT, following approved dosing regimens in patients with various degrees of renal impairment is shown in Figure 7.
Those curves can be used to estimate the time to get to a particular level of recovery, even when the time since the last dose of DABIGATRAN is not precisely known.

Figure 7 Average Time Course for Effects of Dabigatran on aPTT, Following Approved DABIGATRAN Dosing Regimens in Patients with Various Degrees of Renal Impairment
The degree of anticoagulant activity can also be assessed by the Ecarin clotting time ( ECT ). This test actually, may be the most useful one for measuring thrombin inhibition (5), being therefore a more specific measure of the effect of dabigatran than activated partial thromboplastin time ( aPTT ).
PHARMACOKINETICS
Dabigatran etexilate mesylate is absorbed as the Dabigatran etexilate ester. The ester is then hydrolyzed, forming dabigatran, the active moiety. Dabigatran is metabolized to four different acyl glucuronides and both the glucuronides and dabigatran have similar pharmacological activity. Pharmacokinetics described here refer to the sum of dabigatran and its glucuronides. Dabigatran displays dose-proportional pharmacokinetics in healthy subjects and patients in the range of doses from 10 to 400 mg.
ABSORPITION
The absolute bioavailability of dabigatran following oral administration of dabigatran etexilate is approximately 3 to 7%.
It has been discovered that drug’s absorption is better in an acid environment, so dabigatran molecules are bound to a tartaric acid core (5)
Dabigatran etexilate is a substrate of the efflux transporter P-gp.
Cmax occurs at 1 hour post-administration.
Coadministration of the drug with a high-fat meal delays the time to Cmax by approximately 2 hours, but has no effect on the bioavailability of dabigatran; Therefore dabigatran may be administered with or without food.
DISTRIBUTION
Dabigatran is approximately 35% bound to human plasma proteins.
The volume of distribution of dabigatran is 50 to 70 L.
Given twice daily, dabigatran’s accumulation factor is approximately two.
Furthermore, the drug is not able to pass the blood-brain barrier.
METABOLISM
After oral administration, dabigatran etexilate is converted to dabigatran. The cleavage is mediated by ubiquitous esterases hydrolysis to the active principal dabigatran (5)
As Dabigatran is not a substrate, inhibitor, or inducer of CYP450 enzymes, the potential for drug-drug interation is low (5).
Dabigatran is subject to conjugation with glucuronic acid, forming pharmacologically active acyl glucuronides.
ELIMINATION
The half-life of dabigatran in healthy subjects is 12 to 17 hours.
The overall exposure, is increased by 40-60% in healty eldery volounteers primarly due to the physiological reduction of renal clearance and consequently slower dabigatran elimination (5).
Dabigatran is eliminated primarily in the urine; actually renal clearance of Dabigatran is 80% of total clearance after intravenous administration.
The glucuronide conjugated form, is excreted by liver into bile (10)
After oral administration of radiolabeled dabigatran, 7% of radioactivity is recovered in urine and 86% in feces.
| DABIGATRAN ETEXILATE MAIN FEATURES: |
| PHARMACOLOGY |
| Route of administration | Oral |
| Inhibitor type | selective, reversible DTI (Ki= 4.5 nM L-1) |
| Molecular weight | 628 Da (prodrug); 472 Da (active) |
| Prodrug | Double prodrug converted to active form via two intermediates |
| ABSORPTION AND METABOLISM |
| Oral bioavailability | 6–7% (decreased by PPI by 20–25% |
| Involvement of CYP450 | No |
| Food effect | Delayed absorption |
| Plasma protein binding | 35% |
| Metabolism | Occurs in hepatocytes and enterocytes; 20% metabolized to yield activated glucuronide conjugates |
| Elimination | Biliary 20%; Renally 80% |
| PHARMACOKINETICS |
| Pharmacokinetic profile | Linear |
| T max (hr) | 1–3 |
| Half Life | 12–14 |
| CLINICAL CONSIDERATIONS |
| Approved Indications | VTE prophylaxis after hip- or kneereplacement surgery (EU and Canada)
I and II TE Prevention in pt with Non valvular AF |
| Dosage | 150 or 220 mg once daily (VTE prophylaxis)
110 or 150 mg twice daily if pt with AF |
| Dose reduction | Moderate renal impairment (CrCl <50 ml min-1);
contraindicated when CrCl <30 ml min-1) |
| Monitoring | Not needed |
| Potential drug’s interactions | Quinidine, verapamil, amiodarone, rifampicin,
St. John’s wort, strong P-glycoprotein inhibitors |
Table 3: Main characteristics of dabigatran etexilate
| Impact of Renal Impairment on Dabigatran Pharmacokinetics: |
|
| Renal function | CrCl (ml/min) | Increase in AUC | Increase in Cmax | T1/2 (h) |
| Normal | ≥80 | 1x | 1x | 13 |
| Mild | 50-80 | 1.5x | 1.1x | 15 |
| Moderate | 30-50 | 3.2x | 1.7x | 18 |
| Severe | 15-30 | 6.3x | 2.1x | 27 |
Table 4: renal impairment and Dabigatran
Renal Impairment
Exposure to dabigatran increases with severity of renal function impairment (10) (Table 3). Similar findings were observed in the RE-LY trial (11).
Hepatic Impairment
Administration of DABIGATRAN in patients with moderate hepatic impairment showed a large inter-subject variability, but no evidence of a consistent change in exposure or pharmacodynamics.
DRUG INTERACTIONS
As Dabigatran is not a substrate, inhibitor, or inducer of CYP450 enzymes, the potential for drug-drug interation is low (5).
Dabigatran however, is a substrate of P-gp trasporter.
Therefore interaction are possible with drugs that interacts with P-gp.
P-gp INDUCERS
Rifampin: Rifampin 600 mg once daily for 7 days followed by a single dose of dabigatran decreased its AUC and Cmax by 66% and 67%, respectively.
P-gp INHIBITORS
In studies with the P-gp inhibitors ketoconazole, amiodarone, verapamil, and quinidine, the time to peak, terminal half-life, and mean residence time of dabigatran were not affected.
Any observed changes in Cmax and AUC are described below.
DRONEDARONE: Simultaneous administration of dabigatran etexilate and dronedarone (administered once or twice daily) increases exposure to dabigatran by 70 to 140% compared to dabigatran alone.
KETOCONAZOLE: Systemic ketoconazole increased dabigatran AUC and Cmax values by 138% and 135%, respectively, after a single dose of 400 mg, and 153%, and 149%, respectively, after multiple daily doses of 400 mg.
VERAPAMIL: When dabigatran etexilate was coadministered with oral verapamil, the Cmax and AUC of dabigatran were increased. The extent of increase depends on the formulation of verapamil and timing of administration. If verapamil is present in the gut when dabigatran is taken, it will increase exposure to dabigatran with the greatest increase observed when a single dose of immediate-release verapamil is given one hour prior to dabigatran (AUC increased by a factor of 2.4).
AMIODARONE: When dabigatran etexilate was co administered with a single 600 mg oral dose of amiodarone, the dabigatran AUC and Cmax increased by 58% and 50%, respectively. The increase in exposure was mitigated by a 65% increase in the renal clearance of dabigatran in the presence of amiodarone that may persist after amiodarone is discontinued because of the drug’s long half-life. (11).
QUINIDINE: Concomitant quinidine administration (200mg x 5 every 2 hours) increase dabigatran’s AUC and Cmax by 53% and 56%, respectively.
Clarithromycin: Coadministered clarithromycin had no impact on the exposure to dabigatran.
Other Drugs
Clopidogrel: When dabigatran etexilate was given concomitantly with a loading dose of 300 mg or 600 mg clopidogrel, the dabigatran AUC and Cmax increased by approximately 30% and 40%, respectively.
By the way, concomitant administration of dabigatran etexilate and clopidogrel resulted in no further prolongation of capillary bleeding times compared to clopidogrel monotherapy. When comparing combined treatment and the respective mono-treatments, the coagulation measures for dabigatran’s effect (aPTT, ECT, and TT) remained unchanged, and inhibition of platelet aggregation (IPA), a measurement of clopidogrel’s effect, remained unchanged.
Enoxaparin: Enoxaparin 40 mg given subcutaneously for 3 days with the last dose given 24 hours before a single dose of DABIGATRAN had no impact on the exposure to dabigatran or the coagulation measures aPTT, ECT, or TT.
Diclofenac, Ranitidine, and Digoxin: None of these drugs alters exposure to dabigatran.
The concomitant use of proton pump inhibitors(PPI), H2 antagonists, and digoxin did not appreciably change the trough concentration of dabigatran.
PHARMACOGENOMIC
Unfortunately, because of the recent issue of Dabigatran, “no data are still available”:http://www.ncbi.nlm.nih.gov/pubmed/22122181
about the individual difference in response due to various genetic makeups.
This lack of pharmacogenetic data, once fulfilled by currently ongoing studies, could be of great importance as routine monitoring parameters for these agents are not available.
DRUG SAFETY
No prolongations of QTc interval was observed on cardiac electrophysiology studies.
No evidences of hepatotoxicity are emerged on Dabigatran therapy (5)(11) (4)
Increased AST and ALT, are being detected with the same incidence rate between treated patients and control population (4)
SIDE EFFECTS
Most common side effect is bleeding (8)
Nevertheless, if compared with Warfarin, Dabigatran showed a reduction of either major and minor and life treating bleedings incidence (11)(see figure 9), especially when lower 110 mg twice daily dose was administered.
Moreover, as shown in figure 8, Dabigatran therapy is associated with a decrease of minimum 60% of the intracranial bleeding’s incidence, one of most serious complications of Warfarin therapy (11) (10)
Possibly, this could be due to the Dabigatran’s inability to cross the blood-brain barrier .

Figure 8: rate of intracranial bleeding with Dabigatran vs Warfarin in RE-LY trial (4)

Figure 9: Incidence of combined confirmed venous thromboembolism and venous thromboembolism death (A) and of any bleeding in the RE_COVER trial (B) (4)
However, the RE-LY studio, has shown also an unexplained increase in GI bleeding rate in patients treated with 150 mg twice daily Dabigatran if compared with Warfarin-treated ones. (11)
A possible explanation, could be the excess of acidity due to the tartaric acid core (11)
In the months following the approval and commercialization of Dabigatran, there has also been the signalation to FDA, of an excess of bleeding in patients treated with the new drug.
Because of that unexpected finding, FDA made a study to identify the effective truth of those findings and concluded that it was a case of simulated report, possibly due to an initial increased attention and consequently signalling of the Dabigatran’s bleeding side effect (7)
Another common adverse event, is dyspepsia.
It occurred in 11,8% - 11,3% of patients in the RE-LY studio (11), about twice higher than with Warfarin.
Possibly this effect could be due to the tartaric acid core of the drug (11)
Concern has been expressed on rebound coagulation effect in case of cessation of drug’s assumption (4).
Actually, because of the short Dabigatran half-life (12 to 17 hours), patients can rapidly became susceptible to thromboembolism (10)
Because of that, patient’s compliance is a key feature, needed for a safe Dabigatran therapy.
TOXICITY
Main risk of toxicity bound to Dabigatran use, is severe bleeding
If Dabigatran toxicity is important, it could be necessary contrast its action to stop bleeding.
Unfortunately, no antidote exist for DTIs.
The only reverse bleeding agent that could be used, is Recombinant activated FVII (6)
Otherwise, thanks to the poor Dabigatran’s binding to plasma proteins, dialysis could also be considered to solve the acute drug’s toxicity (3)
| COMPARISON OF THE INCIDENCE OF ADVERSE EVENTS DABIGATRAN vs WARFARIN: |
| VARIABLE | DABIGATRAN 110 mg (N=6015) | DABIGATRAN 150 mg (N=6076) | WARFARIN (N=6022) |
| Dyspepsia | 707 (11.8%) | 688 (11.3%) | 348 (5.8%) |
| Dizziness | 486 (8.1%) | 506 (8.3%) | 568 (9.4%) |
| Dispnea | 557 (9.3%) | 580 (9.5%) | 586 (9.7%) |
| Peripheral edema | 473 (7.9%) | 478 (7.9%) | 468 (7.8%) |
| Fatigue | 399 (6.6%) | 401 (6.6%) | 372 (6.2%) |
| Cough | 344 (5.7%) | 348 (5.7%) | 364 (6.0%) |
| Chest Pain | 312 (5.2%) | 377 (6.2%) | 357 (5.9%) |
| Back Pain | 316 (5.3%) | 314 (5.2%) | 337 (5.6%) |
| Arthralgia | 270 (4.5%) | 335 (5.5%) | 346 (5.7%) |
| Nasopharyngitis | 337 (5.6%) | 330 (5.4%) | 336 (5.6%) |
| Diarrhea | 377 (6.3%) | 397 (6.5%) | 346 (5.7%) |
| Atrial Fibrillation | 330 (5.5%) | 357 (5.9%) | 349 (5.8%) |
| Urinary tract infection | 273 (4.5%) | 289 (4.8%) | 335 (5.6%) |
| Upper respiratory tract infection | 288 (4.8%) | 285 (4.7%) | 313 (5.2%) |
| LIVER FUNCTION |
| ALT or AST >3× ULN | 124 (2.1%) | 117 (1.9%) | 132 (2.2%) |
| ALT or AST >3× ULN with concurrent bilirubin >2× ULN | 13 (0.2%) | 13 (0.2%) | 21 (0.3%) |
| Hepatobiliary disorder |
| Serious adverse event | 33 (0.5%) | 34 (0.6%) | 33 (0.5%) |
| Non–serious adverse event | 101 (1.7%) | 109 ( 1.8%) | 112 (1.9%) |
Table 5: Comparison between side effects of Warfarin and Dabigatran (Adapted from the RE-LY study (11) )
NON-CLINICAL TOXICITY
Carcinogenesis, Mutagenesis, Impairment of Fertility
In tests performed on animal models, Dabigatran showed neither mutagenic nor carcinogenic activities.
Moreover in rat fertility study, no adverse effects on male or female fertility were observed, except for a decreased number of implantations in females receiving an equivalent of 3 times of human’s exposure dose.
Bibliography
Katzung. Basic and Clinical Pharmacology, 11th edition
Cochrane Reviews
Linee guida (www.guidelines.gov)
FDA
EMA (European Medicines Agency)
PubMed
New England Journal of Medicine
Cleveland Clinic Journal of Medicine
Nature
Dovepress
Frontiers in Pharmacology
Wikipedia
(1) ELEC;Dabigatran etexilate for the prevention of stroke and systemic embolism in atrial fibrillation.
National Institute for Health and Clinical Excellence (NICE). Dabigatran etexilate for the prevention of stroke and systemic embolism in atrial fibrillation. London (UK): National Institute for Health and Clinical Excellence (NICE); 2012 Mar. 44 p. (Technology appraisal guidance; no. 249).
(2) EMEA/H/C/000829
(3) Guideline for the management of bleeding on Dabigatran (Pradaxa). Portland (ME): Maine Medical Center, Department of Emergency Medicine; 2012 Feb 9. 3 p. [10 references]
(4) Dahl OE. New oral antithrombotics: focus on dabigatran, an oral, reversible direct thrombin inhibitor for the prevention and treatment of venous and arterial thromboembolic disorders. Vasc Health Risk Manag. 2012;8:45-57. PubMed PMID: 22323896; PubMed Central PMCID: PMC3273411.
(5) Lee CJ, Ansell JE. Direct thrombin inhibitors. Br J Clin Pharmacol. 2011 Oct;72(4):581-92. PubMed PMID: 21241354; PubMed Central PMCID: PMC3195735.
(6) O'Dell KM, Igawa D, Hsin J. New oral anticoagulants for atrial fibrillation: a review of clinical trials. Clin Ther. 2012 Apr;34(4):894-901. PubMed PMID: 22417716.
(7) Southworth MR, Reichman ME, Unger EF. Dabigatran and postmarketing reports of bleeding. N Engl J Med. 2013 Apr 4;368(14):1272-4. PubMed PMID: 23484796.
(8) van Ryn J, Goss A, Hauel N, Wienen W, Priepke H, Nar H, Clemens A. The discovery of dabigatran etexilate. Front Pharmacol. 2013;4:12. PubMed PMID: 23408233; PubMed Central PMCID: PMC3569592.
(9) Vanassche T, Verhaegen J, Peetermans WE, VAN Ryn J, Cheng A, Schneewind O, Hoylaerts MF, Verhamme P. Inhibition of staphylothrombin by dabigatran reduces Staphylococcus aureus virulence. J Thromb Haemost. 2011 Dec;9(12):2436-46. PubMed PMID: 22040101.
(10) Wartak SA, Bartholomew JR. Dabigatran: will it change clinical practice?. Cleve Clin J Med. 2011 Oct;78(10):657-64. PubMed PMID: 21968472.
(11) Stuart J. Connolly, M.D., Michael D. Ezekowitz et al Dabigatran versus Warfarin in patients with Atrial Fibrillation. The New England Journal of Medicine 2009 Sept 17, 316(9): 1139-1150. NEJM.org. 10.1056/ NEJMoa0905561
Lavoro svolto da Riccardo Dacomo