DEFINITION
Epilepsy (from the Ancient Greek ἐπιληψία (epilēpsía) — "to seize") is a common chronic neurological disorder characterized by recurrent unprovoked seizures
 Epilepsy
Epilepsy
EPIDEMIOLOGY
age, sex, seasonality, etc
SYMPTOMS
DIAGNOSIS
histopathology
radiology
NMR
laboratory tests
PATHOGENESIS
As a working hypothesis we can assume a major role of GABAergic system as a causal factor of epilepsy. The affected step can be:
glycyrrhetinic acid epilepsy
PATIENT RISK FACTORS
Epilessia, studio Telethon inchioda gli 'aiutanti' dei neuroni
Roma, 14 mag. (Adnkronos Salute) - L'epilessia non si può imputare soltanto ai neuroni: anche altre cellule del cervello, come gli astrociti, hanno un ruolo importante nella genesi delle scariche elettriche tipiche di questa malattia neurologica. Lo dimostra uno studio finanziato da Telethon e dalla Commissione europea, pubblicato sulle pagine di 'Plos Biology'. Il lavoro è il risultato di una collaborazione tra tre diversi gruppi di ricerca legati all'Istituto di neuroscienze del Cnr di Padova e Pisa e all'Istituto neurologico Besta di Milano, coordinati da Giorgio Carmignoto, Gian Michele Ratto e Marco de Curtis. I ricercatori sono riusciti a dimostrare il ruolo attivo nella nascita delle scariche epilettiche degli astrociti, cellule gliali e non neuronali. Sono molto numerose nel cervello dei mammiferi, e dialogano continuamente con i neuroni. Lo studio dimostra come l'interazione tra neuroni e astrociti sia uno dei meccanismi che contribuisce alla generazione delle scariche epilettiche. Ritenuti in passato dei semplici 'aiutanti' dei neuroni, gli astrociti si sono rivelati nel corso del tempo cellule che esercitano nel cervello un ruolo decisamente più attivo. Questo vale evidentemente anche per la genesi delle crisi epilettiche. Monitorando in laboratorio l'attività di neuroni e astrociti, in diversi modelli sperimentali, i ricercatori hanno infatti scoperto che nella zona di generazione delle scariche epilettiche gli astrociti sono in grado di amplificare nei neuroni circostanti lo stato di ipereccitabilità, che può poi tradursi nella scarica epilettica. A riprova di questo, i ricercatori hanno constatato che inibendo l'attività degli astrociti si riducono le scariche epilettiche, e viceversa. Per chi studia l'epilessia, chiarire i meccanismi biologici, tuttora ben poco conosciuti, che portano allo scatenarsi delle crisi epilettiche è fondamentale per poter sviluppare cure. L'epilessia è una patologia cerebrale che può avere basi genetiche oppure essere la conseguenza di malformazioni del cervello, traumi, infezioni, ictus o tumori. Altre volte la causa è addirittura sconosciuta. Qualsiasi sia l'origine, la manifestazione tipica della patologia epilettica è rappresentata sempre da crisi convulsive, più o meno frequenti, che se non opportunamente trattate possono mettere a rischio la sopravvivenza del paziente. Queste crisi sono la conseguenza di un'anomalia nell'attività elettrica dei neuroni, che raggiungono una sorta di ipereccitazione diffusa ed esageratamente sincrona. Ad oggi non esiste una cura risolutiva: l'unico trattamento disponibile è a base di farmaci capaci di arrestare le convulsioni, ma non di eliminare i meccanismi anomali che sono la causa dell'equilibrio alterato del tessuto nervoso. Inoltre, questi farmaci sono inefficaci in circa un terzo dei pazienti che tendono a sviluppare un'epilessia cronica, spesso accompagnata da gravi problemi neurologici e relazionali.Questo studio rappresenta dunque un significativo passo in avanti nella comprensione dei meccanismi cellulari alla base della patologia epilettica e potrebbe aiutare a delineare una nuova strategia terapeutica per l'epilessia che abbia nell'attività degli astrociti il bersaglio principale.
Genetic
Debate: Does genetic information in humans help us treat patients? PRO--genetic information in humans helps us treat patients. CON--genetic information does not help at all. 2008
Epilepsia. 2008 Dec;49 Suppl 9:13-24.
Delgado-Escueta AV, Bourgeois BF.
- PRO: In the past decade, genotyping has started to help the neurologic practitioner treat patients with three types of epilepsy causing mutations, namely (1) SCN1A, a sodium channel gene mutated in Dravet's sporadic severe myoclonic epilepsy of infancy (SMEI and SMEB); (2) laforin (dual specificity protein phosphatase) and malin (ubiquitin E3 ligase) in Lafora progressive myoclonic epilepsy (PME); and (3) cystatin B in Unverricht-Lundborg type of PME. Laforin, malin, and cystatin B are non-ion channel gene mutations that cause PME. Genotyping ensures accurate diagnosis, helps treatment and genetic counseling, psychological and social help for patients and families, and directs families to organizations devoted to finding cures for specific epilepsy diseases. In SCN1A and cystatin B mutations, treatment with sodium channel blockers (phenytoin, carbamazepine, oxcarbazepine, lamotrigine) should be avoided. Because of early and correct diagnosis by genotyping of SCN1A mutations, the avoidance of sodium channel blockers, and aggressive treatment of prolonged convulsive status, there is hope that Dravet's syndrome may not be as severe as observed in all past reports. Genotyping also identifies nonsense mutations in Lafora PME. Nonsense mutations can be corrected by premature stop codon readthrough drugs such as gentamicin. The community practitioner together with epilepsy specialists in PME can work together and acquire gentamicin (Barton-Davis et al., 1999) for "compassionate use" in Lafora PME, a generalized lysosome multiorgan storage disorder that is invariably fatal. In Unverricht-Lundborg PME, new cohorts with genotyped cystatin B mutations have led to the chronic use of antioxidant N-acetylcysteine and combination valproate clobazam or clonazepam plus antimyoclonic drugs topiramate, zonisamide, piracetam, levetiracetam, or brivaracetam. These cohorts have minimal ataxia and no dementia, questioning whether the syndrome is truly progressive. In conclusion, not only is genotyping a prerequisite in the diagnosis of Dravet's syndrome and the progressive myoclonus epilepsies, but it also helps us choose the correct antiepileptic drugs to treat seizures in Dravet's syndrome and Unverricht-Lundborg PME. Genotyping also portends a brighter future, helping us to reassess the true course, severity, and progressive nature of Dravet's syndrome and Unverricht-Lundborg PME and helping us craft a future curative treatment for Dravet's syndrome and Lafora disease. Without the genotyping diagnosis of epilepsy causing mutations we are stuck with imprecise diagnosis and symptomatic treatment of seizures.
- CON: Genotyping of epilepsy may help to better understand the genetics of epilepsy, to establish an etiology in a patient with epilepsy, to provide genetic counseling, and to confirm a clinical diagnosis. However, critical analysis reveals that genotyping does not contribute to an improved treatment for the patients. In order to improve treatment, genotyping would have to (1) improve our ability to select the drug of choice for a given epilepsy or epileptic syndrome; (2) improve our ability to predict the individual risk of adverse reactions to certain drugs; (3) improve our ability to avoid unnecessary treatments or treatments that could aggravate seizures. Many example illustrate the lack of impact of genetic information on the treatment outcome: we do not treat Dravet syndrome more successfully since SCN1A testing became available; we do not treat Lafora disease more successfully since testing for laforin and malin became available; we do not need to know the genetic nature of Unverricht-Lundborg disease or test for the cystatin B mutation in order to select or avoid certain drugs; we do not treat Rett syndrome more successfully since MECP2 testing became available; we do not treat JME more successfully since we know its genetic origin; we do not treat autosomal dominant nocturnal frontal lobe epilepsy more successfully since we know its genetic origin and can test for its mutation. The clinical characteristics as well as the response to treatment of these epilepsy syndromes have been well established before genotyping became available. It can not be argued that genotyping is necessary for establishing a diagnosis or ensure accurate diagnosis. Since not all individuals with given syndromes have been shown to have the corresponding mutation, the clinical diagnosis must have been based on well-established clinical criteria. In addition, the presence or absence of the mutation in a given patient has never been shown to specifically predict the response to any form of treatment, positive or negative. Finally, the appropriate psychological and social help in a given patient will not depend on the identification of a mutation. This does not leave any role for genotyping in epilepsy for the sole reason of improving treatment of the patient. Claiming that the result of genotyping predicts optimal treatment in certain epilepsies is equivalent to stating that genotyping for diabetes has become available and that, based on this breakthrough, insulin can now be selected as the treatment of choice in those who test positive.
Vascular
Genetic
Acquired
Hormonal
Genetic
Acquired
TISSUE SPECIFIC RISK FACTORS
anatomical (due its structure)
vascular (due to the local circulation)
physiopathological (due to tissue function and activity)
COMPLICATIONS
THERAPY
- increasing GABA production
ABSTRACT (The ketogenic diet (KD) is a high-fat, low-carbohydrate, and adequate-protein diet used to treat drug-resistant seizures) 2005
Kirk Nylen, Sergei Likhodii, Peter A. Abdelmalik, Jasper Clarke, and W. McIntyre Burnham
The ketogenic diet (KD) is a high-fat, low-carbohydrate, and adequate-protein diet used to treat drug-resistant seizures. It is currently unknown exactly how the KD exerts its anticonvulsant effects. Animal models are used to study the KD anticonvulsant mechanism of action. In the present study, we explored the ability of two KDs, a 4:1 KD and a 6.3:1 KD (more severe ketosis), to elevate pentylenetetrazole (PTZ) seizure thresholds in adult rats and young rats. When calculating PTZ thresholds, one can use either "absolute latencies" (i.e., the number of seconds until a seizure occurs) or "threshold doses" (i.e., the milligram/kilogram dose of PTZ required to elicit a seizure). When absolute latencies were used, neither KD significantly increased the latency to seizure in adult rats and young rats. Similarly, neither KD elevated threshold doses in adult rats. The 4:1 KD did not elevate threshold doses in young rats; however, the 6.3:1 KD did. Threshold doses are sensitive to differences between group weights, and the largest difference between group weights existed between the 6.3:1 KD group and its respective control group. It remains unclear whether threshold doses, absolute latencies, or some combination of the two methods should be used when determining seizure thresholds in rats fed a KD and seizure tested by using the PTZ infusion test. We conclude that the PTZ-infusion test is not suitable for modeling the anticonvulsant effects of the KD seen clinically, especially when dietary treatments lead to significantly mismatched body weights between the groups.
Neurosteroids Analogs
Neurosteroids and epilepsy. 2010
Curr Opin Neurol. 2010 Apr;23(2):170-6.
Biagini G, Panuccio G, Avoli M.
Dipartimento di Scienze Biomediche, Università di Modena and Reggio Emilia, Modena, Italy.
Abstract
PURPOSE OF REVIEW: Neurosteroids are a family of compounds synthesized directly in the brain by transforming cholesterol into pregnenolone, which is then converted to compounds such as allopregnanolone and allotetrahydrodeoxycorticosterone. In view of their ability to modulate neurotransmission, neurosteroids may influence the clinical course of epileptic disorders. In this review, we highlight two emerging properties of neurosteroids, that is, their anticonvulsant and antiepileptogenic activities. RECENT FINDINGS: It has been shown that fluctuations in neurosteroid synthesis, such as those seen in response to stress or during the ovarian cycle, determine an increase in seizure threshold. Moreover, increased neurosteroid synthesis, presumably occurring in glial cells during epileptogenesis, delays the appearance of recurrent spontaneous seizures in an animal model of temporal lobe epilepsy; such an effect may be due to augmented tonic gamma-aminobutyric acid type A receptor-mediated inhibition. Finally, clinical trials with ganaxolone, an allopregnanolone analogue, have demonstrated beneficial effects in pharmacoresistant epileptic patients, whereas finasteride--which interferes with neurosteroid synthesis - facilitates seizures in catamenial epilepsy. SUMMARY: The overall evidence suggests that neurosteroids may represent a novel therapeutic strategy in epileptic disorders and a future perspective to control epileptogenicity.
Update on the Neurobiology of Alcohol Withdrawal Seizures, 2005
- Abrupt cessation of alcohol intake after prolonged heavy drinking may trigger alcohol withdrawal seizures. Generalized tonic–clonic seizures are the most characteristic and severe type of seizure that occur in this setting. Generalized seizures also occur in rodent models of alcohol withdrawal. In these models, the withdrawal seizures are triggered by neuronal networks in the brainstem, including the inferior colliculus; similar brainstem mechanisms may contribute to alcohol withdrawal seizures in humans. Alcohol causes intoxication through effects on diverse ion channels and neurotransmitter receptors, including GABAA receptors—particularly those containing δ subunits that are localized extrasynaptically and mediate tonic inhibition—and N-methyl-D-aspartate (NMDA) receptors. Alcohol dependence results from compensatory changes during prolonged alcohol exposure, including internalization of GABAA receptors, which allows adaptation to these effects. Withdrawal seizures are believed to reflect unmasking of these changes and may also involve specific withdrawal-induced cellular events, such as rapid increases in α4 subunit–containing GABAA receptors that confer reduced inhibitory function. Optimizing approaches to the prevention of alcohol withdrawal seizures requires an understanding of the distinct neurobiologic mechanisms that underlie these seizures.
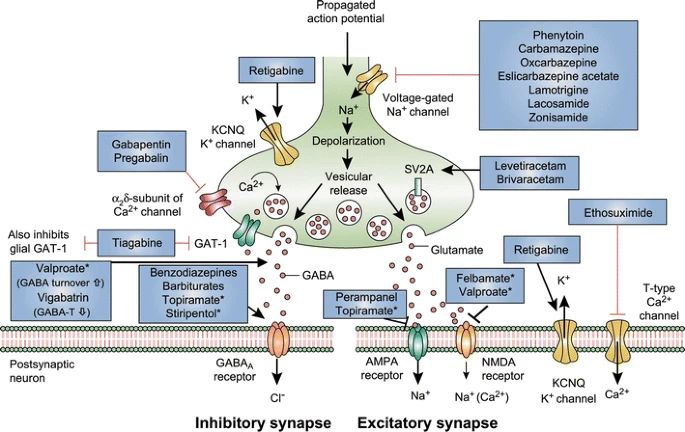
Mechanism of action of clinically approved anti-seizure drugs. Updated and modified from Löscher and Schmidt [151]. Drugs marked with asterisks indicate that these compounds act by multiple mechanims (not all mechanisms shown here). GABA-T GABA aminotransferase, GAT GABA transporter, SV2A synaptic vesicle protein 2A, GABA gamma-aminobutyric acid, NMDA N-methyl-D-aspartate, AMPA α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid, KCNQ a family of voltage-gated potassium channels (also known as the Kv7 family)
Synaptic Vesicle Glycoprotein 2A Ligands in the Treatment of Epilepsy and Beyond, 2018
Brivaracetam: Rationale for discovery and preclinical profile of a selective SV2A ligand for epilepsy treatment, 2016
epilepsy and statin
epilepsy+and+anandamide