DEFINITION
Polycythemia Vera is a chronic myeloproliferative neoplasm of unknown etiology, characterized by abnormal proliferation of all hematopoietic bone marrow elements and an absolute increase in red cell mass and total blood volume, associated frequently with splenomegaly, leukocytosis, and thrombocythemia. Hematopoiesis is also reactive in extramedullary sites (liver and spleen). In time myelofibrosis occurs. The major symptoms are related to hypertension or to vascular abnormalities caused by the increased red cell mass. With currently available treatment, the median survival exceeds 10 years.
Source: CRISP Thesaurus, 2006; Medical Subject Headings, 2011_2011_02_14; NCI Thesaurus, 2010_02D
http://www.diseasesdatabase.com/ddb10302.htm
The disease definition according to a specific consensus conference or to The Diseases Database based on the Unified Medical Language System (NLM)
Polycythemia vera and essential thrombocythemia: 2011 update on diagnosis, risk-stratification, and management, 2011
Janus kinase inhibitors for the treatment of myeloproliferative neoplasias and beyond, 2011
Myeloproliferative neoplasms: molecular pathophysiology, essential clinical understanding, and treatment strategies, 2011
Targeting myeloproliferative neoplasms with JAK inhibitors, 2011
Advances in understanding and management of polycythemia vera, 2011
MeSH PV
Polycythemia Vera (PV) is a myeloproliferative neoplasms (MPN), one of five categories of myeloid malignancies, according to the World Health Organization (WHO) classification system for hematopoietic tumors. ‘‘BCR-ABL1-negative MPN’’ is an operational sub-category of MPN that includes polycythemia vera (PV), essential thrombocythemia (ET), and primary myelofibrosis (PMF). All three disorders are characterized by stem cell-derived clonal myeloproliferation that is skewed towards erythrocytosis in PV and thrombocytosis in ET.

EPIDEMIOLOGY
Polycythemia vera (PV) is a rare disease that affects slightly more men than women. The incidence of PV in the United States per 100,000 population is 2.8 men and 1.3 women.
The mean age at diagnosis is 62 years; it is uncommon in people younger than 30 years. Polycythemia vera (PV) typically affects adult men at a slightly higher rate than women.
SYMPTOMS
Patients with polycythemia vera (PV) can be asymptomatic.
Symptoms of PV can be attributed to different aspects of the disease, including
- Altered blood flow due to high red blood cell mass
- Headaches
- Sweating
- Ringing of the ears
- Blurred vision/blind spots
- Dizziness/vertigo
- Reddish or purplish skin
- Increased platelet levels
- Thrombotic events
- Bleeding (in about 25% of PV patients)
- Angina
- Congestive heart failure
A classic symptom of PV is itching, particularly after exposure to warm water, present in approximately 40% of patients. Gout may be present in up to 20% of patients. A rare but classic symptom of PV is erythromelalgia, characterized by severe burning pain in the hands or feet, usually accompanied by a reddish or bluish coloration of the skin an caused by an increased platelet count or increased platelet "stickiness" (aggregation), resulting in the formation of tiny blood clots in the vessels of the extremity.
Patients with PV are prone to the development of thrombotic events. A major thrombotic complication may sometimes be the first symptom or indication that a person has polycythemia vera.
Headaches, lack of concentration and fatigue are common symptoms that occur in patients with polycythemia vera as well.
DIAGNOSIS
The diagnosis is often suspected on the basis of laboratory tests. Common findings include an elevated hemoglobin level, reflecting the increased number of red blood cells; the platelet count or white blood cell count may also be increased. The erythrocyte sedimentation rate (ESR). Because polycythemia vera results from an essential increase in erythrocyte production, patients have a low erythropoietin (EPO) level.
According to the 2008 WHO diagnostic guidelines, the 2 major diagnostic criteria for polycythemia vera (PV) are:
- Hemoglobin levels greater than 18.5 g/dL in men or 16.5 g/dL in women or other evidence of increased red cell volume
- JAK2V617F mutation status, which is positive in 97% of all patients with PV and therefore strongly supportive of a diagnosis, or other functionally similar mutation, such as the JAK2 exon 12 mutation
However, the absence of a JAK2V617F mutation does not rule out a diagnosis. Thus, for patients with an increased red cell mass and a negative JAK2V617F mutation status, the WHO outlines additional criteria to support a diagnosis of PV.
The laboratory detection of JAK2V617F is highly sensitive (97% sensitivity) and virtually 100% specific for distinguishing PV from other causes of increased hematocrit; the possibility of false-positive or false-negative mutation test result is effectively addressed by the concomitant measurement of serum erythropoietin level, which is expected to be subnormal in more than 85% of patients with PV. A subnormal serum erythropoietin level in the absence of JAK2V617F mandates additional mutational analysis for JAK2 exon 12 mutation in order to capture some of the approximately 3% of patients with PV who are JAK2V617F negative. Bone marrow examination is not essential for the diagnosis of PV because the WHO diagnostic criteria for PV does not require the absence of bone marrow fibrosis.
The 3 minor diagnostic criteria are:
- Bone marrow trilineage myeloproliferation (hypercellularity of erythrocytes, granulocytes and megakaryocytes)
- Subnormal serum EPO levels
- Endogenous Erythroid Colony (EEC) formation in vitro.
A diagnosis of polycythemia vera (PV) requires meeting both major criteria and 1 minor criterion OR 1 major criterion and 2 minor criteria.

PATHOGENESIS
The disease-causing mutation is unknown in PV. However, almost all patients with PV harbor a JAK2 mutation. Other mutations associated with PV involve LNK, CBL, TET2, IDH or EZH2. The pathogenetic relevance of these mutations is currently under investigation but none of them appear to garner the disease specificity or pathogenetic relevance otherwise displayed by BCR-ABL1.

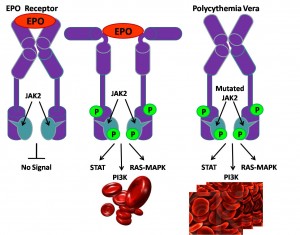
JAK2V617F (Janus kinase 2; 9p24) is the most prevalent mutation in BCR-ABL1-negative MPN, with a mutational frequency of 96% in Polycythemia Vera, 55% in essential thrombocythemia, and 65% in Primary Myelofibrosis.
The presence of JAK2V617F in MPN has been associated with:
- older age,
- higher hemoglobin level,
- leukocytosis,
- lower platelet count.
In PV, a higher mutant allele burden has been associated with pruritus and fibrotic transformation.
JAK2V617F presence or increased allele burden does not appear to affect thrombosis risk, survival or leukemic transformation.
JAK2 exon 12 mutations are relatively specific to JAK2V617F-negative PV (mutational frequency among all PV patients is 3%) and are therefore diagnostically useful in such a setting.
JAK2 exon 12 mutation-positive patients usually present with:
- predominantly erythroid myelopoiesis,
- subnormal serum erythropoietin level,
- younger age at diagnosis.
PATIENT RISK FACTORS
Risk factors for shortened survival in include
- history of thrombosis,
- leukocytosis,
- advanced age,
- anemia.
Leukocytosis has also been associated with leukemic or fibrotic transformation in PV.
Current risk stratification in PV is designed to estimate the likelihood of thrombotic complications:
- Age ≥ 60 years
- history of thrombosis
are the two risk factors used to classify patients with PV into low (zero risk factors) and high (one or two risk factors) risk groups. In addition, because of the potential risk for bleeding, low-risk patients with extreme thrombocytosis (platelet count > 1,000 × 109/L) are considered separately. The presence of cardiovascular risk factors (ie, hypertension, smoking, hypercholesterolemia, diabetes mellitus) is currently not taken under consideration during formal risk categorization.
Risk Stratification In PV
|
- Age>60 years or thrombosis history
|
- Cardiovascular risk factor
|
|---|
| Low | No | No |
| Intermediate | No | Yes |
| High | Yes | / |
Risk of cardiovascular complications from polycythemia vera (PV) is highest in patients 65 years or older and in those with a history of thrombosis.
Whether leukocytosis or a high JAK2V617F mutation allele burden should be included in modeling myeloproliferative neoplasm vascular risk is still under investigation.
COMPLICATIONS
PV may be a risk factor for:
- Myelofibrosis
- Acute myeloid leukemia
- Idiopathic intracranial hypertension
It is important to monitor patients to prevent or treat potential complications of PV. Among the serious risks and complications of PV are thrombotic events and bleeding.
Some patients may progress to myelofibrosis (MF); when this occurs, it is said that a patient has post-PV MF (PPV-MF), which is life-threatening and often fatal. Median survival is about 5.7 years.
A small minority of PV patients can transform to secondary acute myelogenous leukemia (sAML). The prognosis for sAML is much poorer than for de novo AML. sAML is untreatable, and can be rapidly fatal.

THERAPY
Current Treatment Landscape
There are no FDA-approved medicines for polycythemia vera (PV), and the only potential cure is allogeneic stem cell transplant , which is only appropriate for a small subset of high-risk patients. The goals of therapy in PV are, in most instances, adequate prophylaxis against vascular events and, when possible, treatment of symptoms. Therefore, there is an unmet medical need for novel therapies that better address the clinical manifestations of PV.
Some examples of currently used therapies include:
- Aspirin
- Cytoreductive therapy
- Phlebotomy
Patients have a relatively good prognosis with median survival exceeding 15–20 years. Controlled studies have confirmed the antithrombotic value of low-dose aspirin in polycythemia vera (all risk categories). In addition, phlebotomy is indicated in all patients with polycythemia vera with a generally advocated hematocrit target of <45%. Use of hydroxyurea in high-risk polycythemia vera is also recommended.
Polycythemia vera patients who are either intolerant or resistant to hydroxyurea can be treated with interferon-alpha (INF-alpha) (younger patients) or busulfan (older patients). Use of IFN-alpha is limited by its significant adverse event profile (including for pegylated INF-alpha) and its uncertain effects on disease-related complications and survival. Wider use of busulfan has been limited by the fear of leukemogenicity.
Investigational Agents
New treatments are under development and in clinical trials, but have not been FDA-approved for PV. Some of these drugs include
- Cytoreductive therapy
- Histone deacetylase (HDAC) inhibitors
- New immunomodulatory drugs (IMiDs)
- Janus kinase (JAK) inhibitors
Janus Kinase (JAK) inhibitors
Experimental evidence in mouse models and in patient samples suggests that JAK2V617F causes erythrocytosis and progression to myelofibrosis. Furthermore, JAK2V617F allele burden correlates with white blood cell counts and haemoglobin levels in patients with PV. In fact, the ratio of mutant to wild-type JAK2 seems to modulate the phenotypic manifestations of MPNs, with high ratios favouring the development of a PV like phenotype and low ratios inducing an ET like phenotype. Exon 12 JAK2 mutations also result in a myeloproliferative phenotype with associated erythrocytosis and marrow endogenous erythroid colonies in a murine model of retroviral bone marrow transplantation. Hence, mutant JAK2 proteins represent attractive targets for the treatment of MPNs.
"FIRST GENERATION" JAK INHIBITORS
The crucial role of JAK2V617F in the pathogenesis of MPNs spurred the discovery of low molecular mass ATP-competitive compounds to inhibit the activity of this oncoprotein. A small group of four compounds (ruxolitinib, TG101348, lestaurtinib and XL019) with different selectivity for the four members of the JAK family of kinases was first tested in patients with PMF or post-PV or -ET myelofibrosis with intermediate or high risk disease (those with at least one of the following risk factors: age >65 years, constitutional symptoms, haemoglobin <10 g per dL, white blood cell count >25 × 109 per L and/or blood blasts >1%, according to the International Working Group for Myelofibrosis Research and Treatment Prognostic scoring system for PMF).
Ruxolitinib. Ruxolitinib (also known as INCB018424) is an oral, biologically available cyclopentylpropionitrile derivative with potent inhibitory activity against JAK1 and JAK2, moderate activity against TYK2 and negligible activity against JAK3 and a panel of 26 additional kinases. Treatment of Ba/F3 cells expressing JAK2V617F with nanomolar concentrations of ruxolitinib induced a dramatic inhibition of JAK2V617F and STAT5 and ERK1/2 phosphorylation, which was coupled with reduced cellular proliferation and the induction of apoptosis.
Ruxolitinib potently inhibited the proliferation of ex vivo expanded erythroid progenitors obtained from patients with JAK2V617F-positive PV. Furthermore, treatment with ruxolitinib dramati¬cally reduced the levels of the circulating pro-inflammatory cytokines IL-6 and tumour necrosis factor-α (TNFα), which are believed to be responsible for the profound constitutional symptoms that are frequently observed in patients with advanced PMF.
The absorption, distribution, metabolism and excretion of ruxolitinib were determined in healthy volunteers, and the safety and efficacy of oral ruxolitinib were tested in a Phase I/II study involving 153 patients with PMF or post-PV or -ET myelofibrosis. The dose-limiting toxicity (DLT) was grade 4 thrombocytopaenia.
One 28 day cycle of ruxolitinib therapy induced dramatic reductions in multiple fibrogenic, pro-inflammatory and angiogenic growth factors (for example, IL 6 and TNFα) that were markedly elevated before therapy.
One month of ruxolitinib therapy improved total or individual symptom scores (obtained from the Myelofibrosis Symptom Assessment Form) in more than 50% of patients. Response rates were similar between patients regardless of the presence or absence of the JAK2V617F mutation (51% versus 45% respectively) as well as among patients with PMF or post-PV or post-ET myelofibrosis (49% versus 45% versus 62%).
Despite the impressive clinical activity, the JAK2V617F allele burden was only minimally decreased by ruxolitinib (mean maximal suppression of 13% from baseline after 12 cycles of therapy)47. Along with the fact that clinical responses occur both in JAK2V617F-positive and JAK2V617F-negative MPNs, this suggests that ruxolitinib does not selectively target the JAK2V617F mutation. In fact, its effect is more likely to be a result of its promiscuous inhibitory activity over other JAKs, such as JAK1, and the assumed effects on cells carrying mutations in exon 12 of JAK2 or Mpl mutations.
TG101348. TG101348 is an oral ATP-competitive inhibitor that occupies the ATP-binding pocket of JAK2 and inhibits JAK2 and JAK2V617F with higher selectivity compared with other members of the JAK family of kinases. The inhibitory activity of TG101348 was profiled in 223 kinases, with only three, JAK2, FLT3 and RET, being inhibited with IC50 <50 nM. Treatment with TG101348 abrogated the propensity of PV progenitors to differentiate along the erythroid lineage in a dose-dependent manner, with minimal effects on other colony types. TG101348 selectively inhibits erythroid colony formation by JAK2V617F-transduced cord blood progenitors and induces apoptosis in human erythroleukaemia (HEL) and Ba/F3 cells expressing JAK2V617F. TG101348 attenuates the levels of phosphorylated JAK2 and inhibits JAK2 induced phospho STAT5, phospho STAT3, phospho-AKT and phospho ERK1/2 levels both in cultured and in primary human cells obtained from patients with JAK2V617F-positive MPNs.
The remarkable activity shown by TG101348 in preclinical studies led to the launching of a Phase I multicentre study. TG101348 was generally well tolerated, and the DLT was asymptomatic grade 3 or 4 elevations of amylase and lipase, which were reversible. The most frequently reported non-haematological toxicities were diarrhoea (75%), nausea (70%) and vomiting (70%). Grade 4 non-haematological toxicities were not reported. Grade 3–4 neutropaenia and thrombocytopaenia were reported in 15% and 33% of patients, respectively.
Clinical improvement was observed in 49% of patients based on reduction of palpable splenomegaly, which occurred in 56% of patients within 12 weeks and in all cases within 20 weeks of treatment. In addition, 73% of patients with pretreatment leukocytosis had a normalized white blood cell count at the last follow-up visit, which was coupled in many cases with normalization of platelet count. Although most patients improved or experienced resolution of baseline constitutional symptoms, such as pruritus and cachexia, no changes in pro-inflammatory cytokines (IL 2, IL 6, IL 8 and TNFα) were observed during TG101348 therapy. This may be attributable to the higher selectivity of TG101348 for JAK2 relative to JAK1. Of the 22 patients harbouring a JAK2V617F allele burden greater than 20%, 59% experienced a median reduction in mutant allele burden of 60%.
Lestaurtinib. Lestaurtinib (also known as CEP 701) is an orally available multikinase inhibitor with potent and similar activity against wild-type and mutant JAK2V617F. In addition, lestaurtinib is also a potent inhibitor of FLT3, RET and TRKA, among other kinases.
Nanomolar concentrations of lestaurtinib inhibit the growth of cell lines carrying both the wild-type and mutated JAK2 as well as primary erythroid cells from patients with PV, which is coupled with abrogation of JAK2 phosphorylation and downstream inhibition of signalling mediated by STAT5, AKT, ERK, BCL-XL and downregulation of cyclins D1 and D2. Erythroid cells expanded from CD34+ cells from patients with MPNs, but not those from healthy volunteers, were inhibited by lestaurtinib at a concentration of 100 nM. These effects were accompanied by abrogation of STAT5 phosphorylation at clinically achievable concentrations.
Two clinical trials suggest that lestaurtinib is moderately active in myelofibrosis, reducing spleen size in approximately one-third of patients. Preliminary results from a Phase II trial for patients with PV or ET suggest that lestaurtinib, at 80 mg twice daily, may be useful in this setting by induc¬ing reductions in spleen size and haemoglobin levels in a subset of patients.
XL019. XL019 is a highly potent JAK inhibitor with high selectivity against JAK2 over other JAKs. In a Phase I/IIclinical trial, patients with PMF (n = 17) or post-ET or -PV myelofibrosis (n = 13) received treatment with XL019. Most patients (80%) carried the JAK2V617F or the MPLW515L alleles and 37% of the patients who entered the study were transfusion-dependent.
Patients carrying mutations in JAK2 or MPL experienced a reduction in spleen size, in contrast with the absence of response among those carrying wild-type JAK2. Three out of the four patients with accelerated disease (10–19% blasts in the peripheral blood) displayed a marked reduction in blast burden during XL019 therapy. However, 21 (70%) out of 30 patients discontinued XL019 therapy, in some instances owing to nerve conduction abnormalities and/or altered mental status. The unacceptable rate of neurological toxicity has precluded further development of XL019 for the treatment of patients with MPNs.
NOVEL JAK INHIBITORS
The promising preliminary results obtained with first-generation JAK inhibitors stimulated the development of novel JAK inhibitors by several biotechnology companies. These agents are in different stages of development, with some of them (SB1518, AZD1480 and CYT387) already being tested in clinical trials in patients with MPNs and others (INCB16562 and NVP BSK805) still being preclinically characterized.
SB1518. SB1518 is a potent JAK inhibitor with high selectivity against JAK2 and JAK2V617F compared with JAK1 or JAK3. It has proven active against leukaemia cell lines that are dependent on JAK2 activation for their growth; for example, it inhibits the proliferation of Ba/F3 cells that are forced to express JAK2V617F (IC50 140–460 nM).
AZD1480. AZD1480 is a novel potent JAK1 and JAK2 inhibitor. JAK selectivity was evaluated in Ba/F3 cells transduced with a fusion kinase consisting of the JH1 catalytic domain of JAK1, JAK2, JAK3 or TYK2 fused to the oligomerization domain of transcription factor ETV6 (TEL).
CYT387. CYT387 is an aminopyrimidine derivative that preferentially inhibits JAK1 and JAK2 over JAK3. CYT387 therapy restored a normal white blood cell count, haematocrit level and spleen size, and normalized the level of pro-inflammatory cytokines in an MPN mouse model. However, upon CYT387 therapy discontinuation, mice relapsed with JAK2V617F-positive MPNs.
Tasocitinib. Tasocitinib (also known as CP-690550) was initially described as a potent and selective inhibitor of JAK3. Recently it has been shown that this compound inhibits JAK1, JAK2 and JAK3 at similar concentrations. Tasocitinib is a potent inhibitor of immune responses in vitro.
References
Polycythemia vera and essential thrombocythemia: 2011 update on diagnosis, risk-stratification, and management, 2011
Janus kinase inhibitors for the treatment of myeloproliferative neoplasias and beyond, 2011
Myeloproliferative neoplasms: molecular pathophysiology, essential clinical understanding, and treatment strategies, 2011
Targeting myeloproliferative neoplasms with JAK inhibitors, 2011
Advances in understanding and management of polycythemia vera, 2011
neutrophils+and+DHEAs
neutrophils+and+DHEA