Introduction
Autoimmune diseases arise from an inappropriate response of the immune system against substances and tissues normally present in the body.
An always higher number of chronic autoimmune diseases is associated with autoantibodies against central nervous system or peripheral nervous system components, or directed to neuromuscular junction.
The only presence of circulating antibodies is not sufficient to define an autoimmune disease but it is necessary to evaluate their pathogenetic role and autoantibodies with pathogenic role demonstrated are few, for example Acetylcholine Receptor Antibody (AchR-Ab), Voltage Gated Potassium Channel Antibodies (VGKC-Ab), Voltage Gated Calcium Channel Antibodies (VGCC-Ab).
There are also autoimmune diseases associated to cancer, for example Myasthenia Gravis associated to thymoma and Lambert-Eaton Myasthenic Syndrome associated to small cell lung carcinoma, and in these cases autoantibodies are both antineuronal and onconeuronal.
Autoantibodies:
- Antineuronal antibodies:
AChRm-Ab (m=muscular receptor (nicotinic receptor); adult form), MuSK-Ab, Lrp4-Ab, Titin-Ab, RyR-Ab: in Myasthenia Gravis
AChRm-Ab (m=muscular receptor (nicotinic receptor); fetal form): in Arthrogryposis Multiplex Congenital
VGCC-Ab: in Lambert-Eaton Myasthenic Syndrome
VGKC-Ab: in Neuromyotonia
- Onconeural antibodies:
Titin-Ab: in Myasthenia Gravis associated to thymoma
VGCC-Ab: in Lambert-Eaton Myasthenic Syndrome associated to small cell lung carcinoma
The neuromuscular junction (NMJ) has the distinction of being the first site of a defined autoantibody mediated neurological disease
Neuromuscular junction physiology:
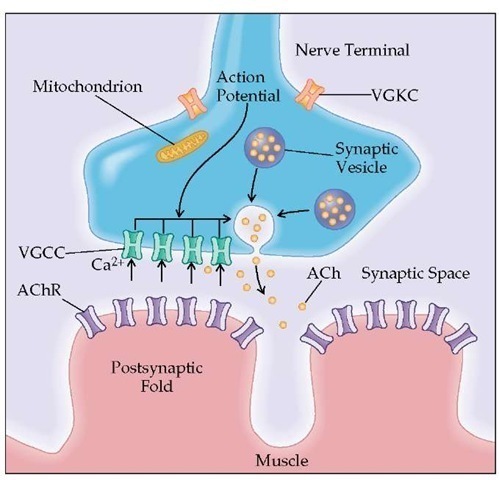
The neuromuscular junction (NMJ) is the communication between nerve and muscle where the electrical nerve impulse is translated into an electrical stimulation to initiate muscle contraction. Acetylcholine (ACh) acts as the chemical messenger between the nerve fibre and the postsynaptic muscle membrane.
Transmission from the nerve ending at the neuromuscular junction takes place at localized sites (active zones). The presynaptic region contains the voltage-gated calcium channels (VGCCs) and synaptic vesicles that hold 6,000 to 8,000 molecules of the neurotransmitter acetylcholine (ACh). When the action potential reaches the nerve terminal, the VGCCs open and Ca2+ flows into the nerve terminal. The entry of Ca2+ triggers the fusion and exocytosis of the synaptic vesicles. When ACh is released into the synaptic space, it rapidly diffuses and binds to the acetylcholine receptor (AChR) located in the end-plate region of the muscle membrane (postsynaptic folds). Activation of AChR by ACh causes ions to flow through AChR across the membrane, initiating the electrical response of the muscle called the end-plate potential (EPP). The EPP spreads from the end-plate region to the surrounding muscle membrane and initiates the impulse response of the muscle that ultimately leads to muscle contraction.
( Disorders of Neuromuscular Transmission )
Neuromuscular junction physiology may be involved by different change on site, cause, clinical features:
- Autoimmmune etiology:
- Myastenia Gravis (MG) (postsynaptic)
- Arthrogryposis Multiplex Congenital (AMC) (postsynaptic)
- Lambert-Eaton Myasthenic Syndrome (LEMS) (presynaptic)
- Neuromyotonia (presynaptic)
- Toxic etiology:
- Cholinesterase inhibitors: Organophosphate Insecticides (Malathion, Parathion)
- Botulinum toxin and alfa-Latrotoxin (presynaptic action)
- alfa-Bungarotoxin and D-Tubocurarine (postsynaptic action)
( Astenia: una maschera per tante identità. Ruolo della patologia della placca neuromuscolare, 2011 )
Autoimmune etiology
Myastenia Gravis:
It is the prototype of the pathology of postsynaptic component of the neuromuscular junction. Myasthenia gravis (MG), the most common autoimmune disease affecting neuromuscular junction, is caused by binding of autoantibodies to molecules involved in neuromuscular transmission (AChRm-Ab, MuSK-Ab, Lrp4-Ab; Titin-Ab and RyanodineR-Ab) and it is characterized by absence of neuromuscular transmission. MG is characterized by muscle weakness that worsens with activity and fluctuates over the course of the day. Ocular symptoms, causing ptosis and diplopia, are the most common presenting symptoms, with around two-thirds of patients progressing to generalized disease, usually within the first 2 years.
Epidemyology:
It is estimated worldwide prevalence between 15 and 179 per million people. The current incidence rates range from 9 to 21 per million population.
The onset of MG is influenced by gender and age. Women are most frequently affected than men. In women MG occurs between 20 and 30 years, in men it occurs between 30 and 40 years; the onset in MG patients with MuSK-Ab occurs predominantly before 40 years, a younger age than MG cases with AChR-Ab.
Diagnosis:
-Clinical history.
-Tensilon Test: this approach requires the intravenous administration of edrophonium chloride or Tensilon, a drug that blocks the degradation (breakdown) of acetylcholine by binding reversibly the enzyme cholinesterase which is inhibited temporarily leading to increases of the levels of acetylcholine at the neuromuscular junction. In people with myasthenia gravis involving the eye muscles, edrophonium chloride will briefly relieve weakness.

According to the Merck Manual, the test is as follows: a syringe is loaded with 10 mg; 2 mg is given intravenous, and if no reaction occurs within 30 sec, the rest is injected. If the patient has myasthenia gravis, muscle function improves suddenly and briefly.
The test can also differentiate between myasthenic crisis and cholinergic crisis (which are for example caused by cholinesterase inhibitors): patients with myasthenic crisis improve; but those with cholinergic crisis worsen (because dangerous cardiorespiratory depression can occur, facilities to maintain respiration and atropine (as an antidote) must be available during the test). In MG patient an obvious increase in strength in weakened muscles strongly suggests the diagnosis of myasthenia gravis. The effect comes on very rapidly, and fades within minutes. In a patient without MG, the Tensilon test will not produce an obvious increase in a previously weak muscle. Some subjective feelings of increased strength are possible but not significant.
-Neurologic examination.
-Electrodiagnostic testing (repetitive nerve stimulation): repetitive nerve stimulation studies demonstrate a rapid reduction (> 12%) in the amplitude of the MAP (=muscle action potential).
-Presence into the serum of antibodies directed to specific proteins in the neuromuscular junction.
-Computed tomography (CT) or magnetic resonance imaging (MRI) may be used to identify an abnormal thymus gland or the presence of a thymoma.
Antibodies:
MG patients can be divided in:
- AchRm-Ab+ (m=muscular receptor) (80-85%) with antibodies directed to nicotinic acetylcholine receptor (AChR) of the skeletral muscle;
- AchRm-Ab- or AchRm seronegative MG patients (15-20%); they can be divided into two groups:
-- MuSK-Ab MG patients (40%): 8%-10% of patients with generalized disease have antibodies to muscle specific tyrosine kinase receptor (MuSK)
-- AchRm-Ab/MuSK-Ab seronegative MG patients (60%)
AchRm-Ab: Acetylcholine Receptor Antibodies
It is now known that antibodies directed against NMJ are present in the serum of patients with this disease, and that AchRm antibody is directly implicated in the disturbance of nicotinic acetylcholine receptor (AChR) function which underlies the disorder of neuromuscular transmission.
AChRm-Ab are found in 85-90% of the patients with generalized disease and in 75% of the patients with restricted ocular myasthenia.
These antibodies are directed against the nicotinic AChR.
The nicotinic AChR is a multimeric protein comprised in adults of two α subunits and one each of β, δ, and ε subunits. Each α subunit has a binding site for Ach.
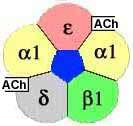
Principally the antibodies are directed to α1-subunit of AChR on an extracellular site called ” MIR“ (=main immunogenic region); other antibodies are directed to ε-subunit.

It has been shown that IgG is present on the postsynaptic membrane, and that its distribution follows that of AChR. There is high heterogeneity of MG anti-AChR and it has confirmed that the antibody is polyclonal in most patients.
Components of the complement system (C3 and C9)have also been shown to be present.
IgG, C3 and C9 lead to a destructive autoimmune process in the postsynaptic membrane with characteristic morphological changes at the endplate.
Ultrastructural changes in the neuromuscular junction: reduction of AchR; flattening of the junctional folds; widening of the synaptic space.

Methodology: AChR antibody testing is done using direct and indirect enzyme linked immunosorbent assay (ELISA) technique.
MuSK-Ab: Muscle Specific tyrosine Kinase Antibodies
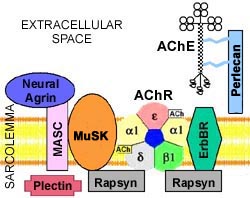
Muscle-specific tyrosine kinase (MuSK) is an AChR associated protein expressed in the mature NMJ and involved in clustering of AChR.
MuSK is a transmembrane polypeptide selectively expressed in skeletral muscle. It is part of the agrinreceptor, an essential protein in building the neuromuscular synapses. Experimental data show that agrin-induced activation of MuSK by tyrosine phosphorylation results in aggregation and expression of specific muscle proteins, AChR included.
About half of AchRm-Ab seronegative MG cases has antibodies against a muscle-specific tyrosine-kinase protein (MuSK). MuSK-Ab may bind the external domain.
Bound complement, C3, was rarely detected at the endplates of MuSK-Ab MG patients because MuSK antibodies are predominantly immunoglobulin IG4 – a subclass that does not activate complement.
Methodology: MuSK antibodies can be detected using a sensitive and highly specific radioimmunoassay (RIA Immunoprecipitation)
Ultrastructural changes in the neuromuscular junction: studies show that MuSK-Ab cause a 20% decrease of AChR number on the surface of the postsynaptic membrane so there is a cluster dispersion of AchR.
Patients with MuSK antibodies are more often women, and can have distinctive clinical patterns of disease including an oculopharyngeal pattern and neck, shoulder, and respiratory weakness pattern.
( Myasthenia gravis Five new things, 2013 )
In relation to muscle ultrastructure histopathological investigation it has been shown that the most common changes in AChR-Ab MG patients are fiber atrophy, myofibrillar disarray, and Z-line streaming; and that mitochondrial abnormalities seem to be more prominent in MuSK-Ab MG patients: mitochondria are giant, swollen, and degenerated with fragmented cristae.
( Myasthenia Gravis with Anti-MuSK Antibodies: Clinical Features and Histopathological Changes, 2012 )
Lrp4-Ab: lipoprotein receptor related protein 4 Antibodies

Antibody testing for AChR and MuSK are confirmatory in MG, but around 10% of patients remains antibody-negative. A protein recently has generated a lot of interest, as antibodies to this protein called low-density lipoprotein receptor–related protein 4 (Lrp4), are believed to be found in a proportion of AchRm/MuSK seronegative MG patients (so double negative: negative for both AChR and MuSK antibodies).
Lrp4 is required for agrin-induced activation of MuSK: agrin interacts with Lrp4 to increase its interaction with Musk and the dimerization of Musk and thus Musk activation. 20 antibodies to Lrp4 were first identified in 2011, and 3%–50% of double-seronegative have Lrp4 antibodies. In addition to patients with double-seronegative MG, Lrp4 antibodies can be detected at low frequency in MuSK-positive patients and rarely in patients with other neurologic diseases (including patients with neuromyelitis optica), but have not yet been found in AChR-positive patients. A portion of sera from Lrp4 antibody-positive patients in cultured myotubes disrupted the agrin-Lrp4 interaction leading to the inhibition of agrin induced AChR aggregation, suggesting a possible pathogenic role for the antibodies. While testing for AChR and MuSK antibodies remains the mainstay of diagnostic testing in MG and detects antibodies in around 90% of patients with generalized MG, testing for Lrp4 may increase the sensitivity of MG antibody testing to 95% in the near future.
( Myasthenia gravis Five new things, 2013 )
Titin-Ab and RyR-Ab: Titin Antibodies and Ryanodine Receptor Antibodies
30% of sera of patients with myasthenia gravis (MG), contains Titin and Ryanodine Receptor (RyR) antibodies. The presence of Titin-Ab and RyR-Ab is associated with more severe disease of MG: Titin-Ab may affect the contractile machinery of myofibers; RyR-Ab may impair excitation-contraction with contribute to muscle weakness in MG patients.


In early-onset MG (before 50 years of age), the presence of Titin-Ab strongly suggests a thymoma and Titin-Ab may be used as tumor markers.
( Titin and ryanodine receptor antibodies and neuromuscular involvement in myasthenia gravis, 2008 )
The role of thymus:
The role of thymus in the pathogenesis of myasthenia gravis is well-known. The thymus is a critical organ for T-cell education and elimination of auto-reactive T-cells and it is the central organ for immunological self-tolerance.
The thymus plays a major role in MG. Thymic abnormalities are frequently present in MG, including hyperplasia in about 65% of cases and thymoma in 10%. The thymus contains myoid cells that express the AChR antigen, antigen presenting cells, and immunocompetent T-cells. MG initiates into the thymus by immunogenic presentation of locally produced nicotinic acetylcholine receptor (AChR) to potentially autoimmune T cells. The inflammatory environment within the MG thymus, contribute to the induction and maintenance of the anti-AChR autoimmune response. Thymic abnormalities cause the breakdown in tolerance that causes an immune-mediated attack on AChR in myasthenia gravis. Thymus pathology in MuSK-positive MG patients is not so evident because of the relatively minor incidence of this subgroup in the population of myasthenic patients.
Histopathological studies often demonstrate thymic hyperplasia in which the changes tipically seen are lymphoid follicles with germinal centres in AChR-Ab positive MG patients. In contrast, in the thymuses from MuSK-Ab positive MG patients these features are significantly less than in the AChR-Ab patients: thymus changes in these cases resemble those in normal aging.

( Myasthenia Gravis with Anti-MuSK Antibodies: Clinical Features and Histopathological Changes, 2012 )
Myasthenia Gravis treatment:
1) Improve neuromuscular transmission: Cholinesterase inhibitors
2) Inhibit the autoimmune process:
- immunosuppressive therapies: corticosteroids; immunosuppressants; biological drugs
- immunomodulatory therapies: plasmapheresis and intravenous immunoglobulins
3) Modify the natural history of the disease: thymectomy
1) Cholinesterase inhibitors: they are drugs that keep Ach in the NMJ for a longer time inhibiting its degradation by Cholinesterasi enzyme. The first known cholinesterase inhibitor was PHYSOSTIGMINE, a natural alkaloid isolated from the poisonous Calabar beans of the West African vine Physostigma venenosum.

Physostigmine has been used in the treatment of myasthenia gravis since the 1930.
( Walker MB: Treatment of myasthenia gravis with physostigmine. Lancet 1934;1:1200-1201 )
In 1935, Percy L. Julian (1899-1975) first synthesized the drug physostigmine.
Cholinesterase inhibitors provide temporary relief from symptoms but do not alter the disease course.
2a) Immunosuppressive therapies: most patients respond well to immunosuppressive therapies. The mainstay of immunosuppressive treatment for MG is prednisone. In addition immunosuppressants such as azathioprine, cyclosporine, mycophenolate mofetil, tacrolimus, have also been shown to be effective. Despite this, some patients with MG remain refractory to treatment. Biological drugs are Rituximab and Etanercept ( “Neuromuscolar junction autoimmune disease: muscle specific kinase antibodies and treatments for myasthenia gravis, 2005":http://www.ncbi.nlm.nih.gov/pubmed/16155434 )
-- Rituximab: Rituximab is a monoclonal antibody that induces complement-dependent cytotoxicity against CD20 positive plasma cells leading to a B-lymphocyte depletion. Rituximab has shown to be effective in other antibody-mediated autoimmune disorders such as systemic lupus erythematosus, dermatomyositis, chronic inflammatory demyelinating polyneuropathy and immunoglobulin M associated polyneuropathies.
-- Etanercept: Etanercept is a recombinant human tumour necrosis factor receptor:Fc that inhibits tumour necrosis factor-alfa, which is one of the cytokines that has been implicated in the pathogenesis of some autoimmune diseases, but it acts to dysregulate cytokine networks and can cause exacerbation in some patients so this treatment is only used used in specialized centres, which can perform cytokine evaluation.
2b) Immunomodulatory therapies: for patients in myasthenic crisis, with severe disease not responding to immunosuppressive therapy, or preparing for thymectomy, plasmapheresis (PLEX) and intravenous immunoglobulins (IVIg) are used to achieve rapid improvement. However there is little information about which treatment strategy is better. The American Academy of Neurology (AAN) published guidelines for the use of IVIg in the treatment of neuromuscular disorders, and recommended IVIg should be considered in the treatment of MG (level B); more controversially, consensus guidelines on the use of PLEX in neurologic disease published by the AAN in 2011 suggested there is insufficient evidence for the use of PLEX in acute MG crisis, due to the lack of randomized trials.
In clinical practice IVIg and PLEX are equally used for treatment of severe MG or MG exacerbations, but PLEX seems superior in true crisis requiring ventilator support. IVIg is easy to administer and generally well-tolerated, but may require longer for a clinically meaningful effect than PLEX. PLEX generally requires 10 days of therapy if treatments are given every other day.
Finally, a cost-benefit analysis of IVIg vs PLEX concluded that IVIg is associated with lower hospital costs.
3) Thymectomy: While thymectomy is considered standard treatment for the 10%–20% of MG cases with thymoma (the first thymectomy for thymomatous MG occurred over 70 years ago), the role of thymectomy in nonthymomatous MG is not clear (and it is generally not considered appropriate for purely ocular MG). Infact in these MG patients: thymectomy often results in clinical improvement in AChR-Ab positive patients; the role of thymectomy in MuSK-Ab positive MG remains uncertain: recent studies reported no reduction in MuSK antibody titles following thymectomy with no difference in clinical status between MuSK-Ab positive MG patients with or without thymectomy.
( Myasthenia gravis Five new things, 2013 )
Arthrogryposis Multiplex Congenital (AMC):
It is an autoimmune congenital disease caused by AchRm antibodies in which the transfer of these antibodies is from a woman to her baby. This is because the woman's antibodies can cross the placenta and affect the baby resulting in 'neonatal' MG, a condition that usually only lasts for days or weeks and seldom has any long-term consequences for the baby. However, there are a few reports of babies suffering from a more severe condition, involving complete paralysis and resulting in permanent fixed joints and other deformities.
Antibodies are directed to γ-subunit in the fetal form of the acetylcholine receptor.

( Acetylcholine Receptors in the Fetus and a Rare Form of Myasthenia, 2000 )
Pathology of presynaptic component of the neuromuscular junction:

- Lambert-Eaton Myasthenic Syndrome (LEMS) with VGCC-Ab: Voltage Gated Calcium Channel Antibodies
Lambert–Eaton Myasthenic Syndrome is a prototype of the pathology of pre-synaptic component of the neuromuscular junction in which the reduced neuromuscular transmission depends on defect in the release of Ach. It is a rare autoimmune disorder that is characterised by muscle weakness of the limbs. It is the result of an autoimmune reaction in which antibodies are formed against presynaptic Voltage Gated Calcium Channels in the NMJ. Voltage-dependent Calcium Channels are a group of voltage gated ion channels found in excitable cells with a permeability to the ion Ca2+. These channels are slightly permeable to sodium ions, so they are also called Ca2+-Na+ channels, but their permeability to calcium is about 1000-fold greater than to sodium under normal physiological conditions. At physiologic or resting membrane potential, VDCCs are normally closed. They are activated (opened) at depolarized membrane potentials and that is the source of the "voltage-dependent" epithet. Activation of particular VDCCs allows Ca2+ entry into the cell, which results in release of neurotransmitter Ach.
The VGCCs in LEMS are no longer in parallel rows but are aggregated as a result of cross-linking by the specific IgG antibodies. Complement is not involved in the process
People who develop LEMS are usually over 40, although it may occur at any age. Around 60% of those with LEMS have an underlying malignancy, most commonly small cell lung cancer.
The diagnosis is usually confirmed with electrodiagnostic testing and blood test.
If the disease is associated with cancer, direct treatment of the cancer often relieves the symptoms of LEMS.
Other treatments often used are steroids, azathioprine and intravenous immunoglobulin, which suppress the immune system, and pyridostigmine and 3,4-diaminopyridine, which enhance the neuromuscular transmission. Occasionally, plasma exchange is required to remove the antibodies.
- Neuromyotonia with VGKC-Ab: Voltage Gated Potassium Channel Antibodies
Neuromyotonia is a rare autoimmune disease presenting as isolated Neuromyotonia or in association for example with MG. Patients with Neuromyotonia have myotonia, and often muscle hypertrophy. Other patients have cramp-fasciculation syndrome and peripheral neuropathy.
Affected patients have antibodies against presynaptic Voltage Gated Potassium Channels in the NMJ. Neuromyotonia is generated by the opening of the VGCCs: inactivation of the VGKCs due to VGKC-Ab leads to a prolonged depolarization of the nerve terminal, prompting the calcium channel to remain open longer, so more calcium enters into the terminal of the nerve and more quanta of Ach are released.
This autoimmune disorder responds to plasma exchange, intravenous immune globulin, or immunosuppressants.
Toxic etiology
Cholinesterase inhibitors: Organophosphate Insecticides (Malathion, Parathion)
Cholinesterase inhibitors lead to an accumulation of acetylcholine that causes continuous stimulation of the muscle. Organophosphate Insecticides (Malathion, Parathion) give paralysis by binding irreversibly the enzyme which is inhibited semi-permanently.
Botulinum Toxin:

Target molecules of botulinum (BoNT) and tetanus (TeNT) toxins inside the axon terminal.
Botulinum toxin is a protein and neurotoxin produced by the bacterium Clostridium botulinum. The toxin must get inside the axon terminals to cause paralysis. The toxin can be taken into neurons by endocytosis. Eight proteins are known. They inhibit the release of Ach.
The type A toxin proteolytically degrades the SNAP-25 protein, a type of SNARE protein. The SNAP-25 protein is required for vesicle fusion that releases neurotransmitters from the axon endings (in particular acetylcholine). Botulinum toxin specifically cleaves these SNAREs, so prevents neurosecretory vesicles from docking/fusing with the nerve synapse plasma membrane and releasing their neurotransmitters.
( Botulinum toxin )
alfa-Latrotoxin, Black widow spider venom:

Binds selectively to presynaptic receptors, induces massive quantal release of ACh, fusion of synaptic vesicles, with hyperactivation of motor and autonomic PNS cholinergic synapses most notably NMJ.
The following mechanism is suggested for receptor-mediated effects. Three receptors for α-latrotoxin have been described: neurexin; latrophilin (aka CIRL, Calcium-Independent Receptor for Latrophilin); protein tyrosine phosphatase sigma (PTPσ).
The toxin stimulates a receptor, most likely latrophilin, which is a G-protein coupled receptor linked to Gαq/11. The downstream effector of Gαq/11 is phospholipase C (PLC).When activated PLC increases the cytosolic concentration of IP3, which in turn induces release of Ca2+ from intracellular stores. This rise in cytosolic Ca2+ may increase the probability of release and the rate of spontaneous exocytosi. Latrophilin with α-LTX can induce the activation of Protein Kinase C (PKC). PKC is responsible for the phosphorylation of SNARE proteins. Thus latrophilin with α-LTX induces the effect of exocytosis of transport vesicles. The exact mechanism has to be discovered.
( alpha-Latrotoxin )
alfa-Bungarotoxin:

α-Bungarotoxin (α-BTX) is one of the bungarotoxins, components of the venom of the elapid snake Taiwanese. It is a neurotoxic protein that is known to bind irreversibly and competitively to the nicotinic acetylcholine receptor found at the neuromuscular junction, causing paralysis, respiratory failure and death in the victim. The toxic, used with radioactive markers, shows depletion of the membranes post-synaptic receptor.
( alpha-Bungarotoxin )
D-tubocurarine:

Tubocurarine (red), Acetylcholine (blue), Acetylcholinesterase (yellow)
D-tubocurarine is the active ingredient of the curare that results from the bark of the roots of poisonous plants. D-tubocurarine is a blocking and non-depolarizing agent of the neuromuscular junction. It binds irreversibly to receptors and makes them inactive.