DEFINITION
Sickle Cell Disease. is an inherited genetic disorder characterized by a mutation in the beta-globin gene HBB.
The defective hemoglobin causes an abnormal, rigid, sickle shape of red blood cells at low pO2.

This abnormality can result in periodic painful episodes, chronic anemia, serious infections and damage to body organs.
People with the disease are born with two sickle cell genes, one from each parent. If you only have one sickle cell gene, it's called sickle cell trait. and doesn’t present symptomatology.
Other, rarer forms of sickle-cell disease include sickle-haemoglobin C disease (HbSC), sickle beta-plus-thalassaemia (HbS/beta+) and sickle beta-zero-thalassaemia (HbS/beta0).
EPIDEMIOLOGY
Sickle cell anemia affects millions of people throughout the world, especially people of sub-Saharan Africa, the Caribbean, the Middle East, the Indian subcontinent and Mediterranean countries.
Although the disease is endemic only in certain parts of the, greater population mobility makes recognition of this disease of paramount importance in surgical practice.
PATHOGENESIS
A point mutation in the beta-globin chain of haemoglobin causes the substitution of the glutamic acid with the hydrophobic aminoacid valine at the sixth position.
The beta-globin gene is found on the short arm of chromosome 11.
The association of two wild-type beta-globin subunits with two mutant beta-globin subunits forms haemoglobin S (HbS).
Sickle trait provides a survival advantage over people with normal hemoglobin in regions where malaria is endemic.
Sickle cell trait provides neither absolute protection nor invulnerability to the disease.
Rather, people (and particularly children) infected with P. falciparum are more likely to survive the acute illness if they have sickle cell trait.
Sickle trait red cells infected with the P. falciparum parasite deform, presumably because the parasite reduces the oxygen tension within the erythrocytes to very low levels as it carries out its metabolism.
Deformation of sickle trait erythrocytes would mark these cells as abnormal and target them for destruction by phagocytes
Since sickle cells are removed from the circulation and destroyed in the reticuloendothelial system, selective sickling of infected sickle trait red cells would reduce the parasite burden in people with sickle trait.
These people would be more likely to survive acute malarial infections.
When these people with sickle cell trait procreate, both the gene for normal hemoglobin and that for sickle hemoglobin are transmitted into the next generation
CELLULAR FUNCTIONS
biological function

The normal Hb does not show any hysteretic behavior that would impair transport by slowing uptake in the lung and release in the peripheral tissues (left panel).
In the case of Hb S the dissociation curve shows a hysteretic behavior that dramatically impairs the release to the tissues, but avoids ischemia (right panel).
Δabs is the percentage of deoxygenated Hb.
it demonstrates that HbS has much affinity with oxigen then the normal Hb and so it has more difficult to release oxigen to the tissues.
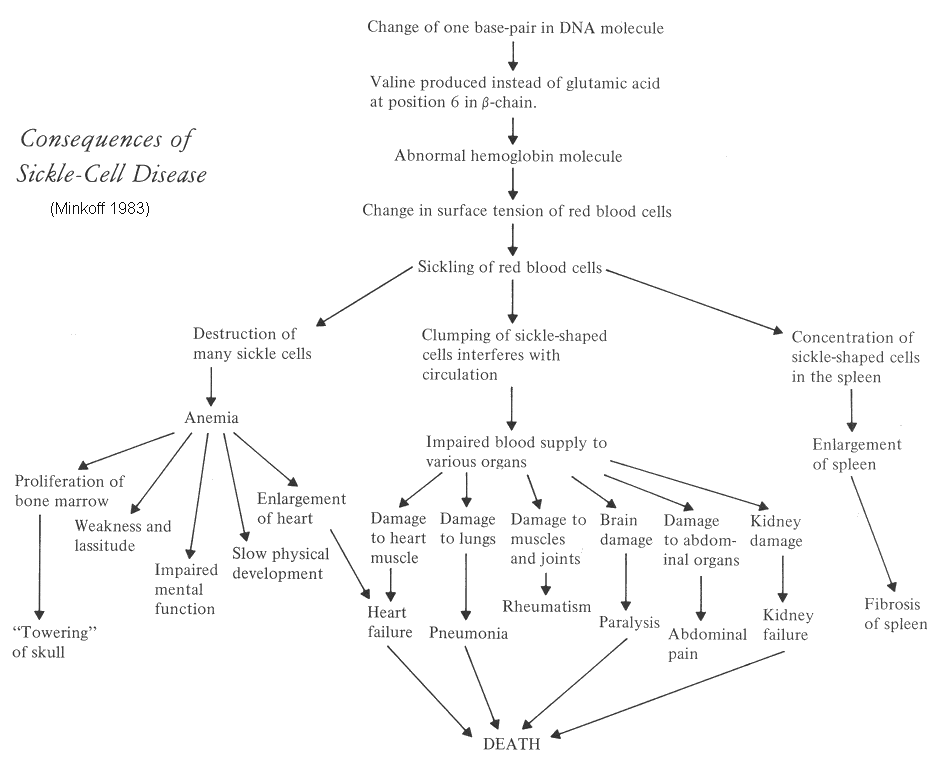
Under low-oxygen conditions, the absence of a polar amino acid at position six of the beta-globin chain promotes the non-covalent polymerisation of haemoglobin, which distorts red blood cells into a sickle shape and decreases their elasticity.
The loss of red blood cell elasticity is central to the pathophysiology of sickle-cell disease.
Normal red blood cells are quite elastic, which allows the cells to deform to pass through capillaries.
In sickle-cell disease, low-oxygen tension promotes red blood cell sickling and repeated episodes of sickling damage the cell membrane infact the adherence of sickle cells to vascular endothelium results in intimal hyperplasia in larger vessels and it decrease the cell's elasticity.

These cells fail to return to normal shape when normal oxygen tension is restored. As a consequence, these rigid blood cells are unable to deform as they pass through narrow capillaries, leading to vessel occlusion and ischaemia.
The actual anemia of the illness is caused by hemolysis, the destruction of the red cells inside the spleen, because of their misshape. Although the bone marrow attempts to compensate by creating new red cells, it does not match the rate of destruction. Healthy red blood cells typically live 90-120 days, but sickle cells only survive 10–20 days.
The high white blood cell count found in most patients with sickle cell anemia results in the production of injurious cytokines.
Known and unknown environmental factors can precipitate vaso-occlusion. Acidosis in muscle or body fluids, dehydration, cold temperatures and infections are known precipitants.
Patients with sickle cell disease can adapt to a hematocrit that is one half the normal value.
SYMPTOMS
The clinical course of sickle cell anemia does not follow a single pattern: some patients have mild symptoms and some have very severe symptoms.
The basic problem, however, is the same: the sickle-shaped red blood cells tend to get stuck in narrow blood vessels, blocking the flow of blood that results in a vaso-occlusive crisis.
This results in the following conditions:
- Hand-foot syndrome.
When small blood vessels in hands or feet are blocked, pain and swelling can result, along with fever.
This may be the first symptom of sickle cell anemia in infants as early as six months of age and may occur in children with sickle trait.
- Pain that occurs unpredictably in any body organ or joint (arms, legs, chest and abdomen).
A patient may experience pain wherever sickled blood cells block oxygen flow to tissues.
Some patients have painful episodes (also called crises) less than once a year, and some have as many as 15 or more episodes in a year.
Sometimes pain lasts only a few hours; sometimes it lasts several weeks.
For especially severe ongoing pain, the patient may be hospitalized and treated with painkillers and intravenous fluids. Pain is the principal symptom of sickle cell anemia in both children and adults.
- The spleen is frequently affected.
It is usually infarcted before the end of childhood in individuals suffering from sickle-cell anaemia. So it increases the risk of infection from encapsulated organisms; preventive antibiotics and vaccinations are recommended for those with such asplenia.
- Eye problems.
The retina can deteriorate when it does not get enough nourishment from circulating red blood cells.
It can be serious enough to cause blindness.
- Acute papillary necrosis in the kidneys.
- Leg ulcers.
- Priapism. and infarction of the penis.
Besides sickle cells are destroyed rapidly in the organism causing:
- Chronic anemia can be manifested with fatigue, paleness, shortness of breath and then with delayed growth and puberty in children and often a slight build in adults, but also may not be significant anemia.
- Jaundice.
- Cholelithiasis (gallstones) and cholecystitis
COMPLICATIONS
- Acutely worsening the patient's baseline anaemia with:
- Parvovirus B19. infection nearly completely prevents red blood cell production for two to three days, causing aplastic crises with pallor, tachycardia, and fatigue.
- Splenic sequestration crises. Acute, painful enlargements of the spleen that involve an abdomen bloated and very hard.
- Hemolytic crises, particularly common in patients with co-existent G6PD deficiency

- Acute chest syndrome is a condition, similar to pneumonia, characterised by fever, chest pain, difficulty breathing, and pulmonary infiltrate on a chest X-ray. It can be triggered by painful crisis, respiratory infection, bone-marrow embolisation, or possibly by atelectasis, opiate administration, or surgery
- Stroke. The defective hemoglobin damages the walls of the red blood cells, causing them to stick to blood vessel walls. This can result in the development of narrowed, or blocked, small blood vessels in the brain, causing a serious, life-threatening stroke. Cerebral infarction occurs in children, and cerebral hemorrhage in adults.
- Infections. They are more vulnerable to infections and have a harder time fighting them off once they start. This is the result of the functional asplenia. Infants and young children, especially, are susceptible to bacterial infections (encapsulated organisms such as Streptococcus pneumoniae and Haemophilus influenzae) that can kill them in as little as 9 hours from onset of fever. Pneumococcal infections used to be the principal cause of death.
- Pulmonary hypertension leading to strain on the right ventricle and a risk of heart failure; typical symptoms are shortness of breath, decreased exercise tolerance and episodes of syncope.
- Chronic renal failure: hypertension, proteinuria, hematuria and worsened anaemia. If it progresses to end-stage renal failure, it carries a poor prognosis.
- During pregnancy, intrauterine growth retardation, spontaneous abortion and pre-eclampsia.
DIAGNOSIS
Early diagnosis of sickle cell anemia is very important.
Children who have the disease need prompt and proper treatment.
More than 40 states now perform a simple, inexpensive blood test for sickle cell disease on all newborn infants.
This test is performed at the same time and from the same blood samples as other routine newborn screening tests.
It's also possible for doctors to diagnose sickle cell anemia before birth.
This is done using a sample of amniotic fluid or tissue taken from the placenta or if both parents are accessible, studies of parental blood can aid in the diagnosis of sickle cell disease in the child. DNA analysis provides the most accurate diagnosis in patients of any age, but it is still relatively expensive.
In newborns, as well as older persons, can be diagnosed by hemoglobin electrophoresis., isoelectric focusing, high-performance liquid chromatography (HPLC.) or DNA analysis. In general, these tests have comparable accuracy.
However hemoglobin electrophoresis is the most widely used diagnostic test that detect abnormal haemoglobin forms.
It is a form of gel electrophoresis on which the various types of haemoglobin move at varying speeds. Sickle-cell haemoglobin (HgbS) and haemoglobin C with sickling (HgbSC)—the two most common forms—can be identified from there.
The diagnosis can be confirmed with high-performance liquid chromatography.
Genetic testing is rarely performed, as other investigations are highly specific for HbS and HbC.
These tests also tell whether the child carries the sickle cell trait.
Besides the full blood count reveals haemoglobin levels in the range of 6–8 g/dL with a high reticulocyte count.
A blood film may show features of hyposplenism (target cells and Howell-Jolly bodies) and can be induced sickling of the red blood cells by the addition of sodium metabisulfite. The presence of sickle haemoglobin can also be demonstrated with the "sickle solubility test".

A mixture of haemoglobin S (Hb S) in a reducing solution (such as sodium dithionite) gives a turbid appearance, whereas normal Hb gives a clear solution.
An acute sickle-cell crisis is often precipitated by infection. Therefore, a urinalysis to detect an occult urinary tract infection, and chest X-ray to look for occult pneumonia should be routinely performed.
To prevent the complications patients need routinary transcranial doppler, cerebral angioRMN, abdominal echography and bone densitometry.
TREATMENTS
Proper nutrition, good hygiene, bed rest, protection against infections, and avoidance of other stresses all are important in maintaining good health and preventing complications. Regular visits to a physician or clinic that provides comprehensive care are necessary to identify early changes in the patient's health and ensure immediate treatment.
Today, with good health care, many people with sickle cell anemia are in reasonably good health much of the time and living productive lives. In fact, in the past 30 years, the life expectancy of people with sickle cell anemia has increased.
- Blood Transfusions
Transfusions correct anemia by increasing the number of normal red blood cells in circulation. They can also be used to treat spleen enlargement in children before the condition becomes life-threatening. Regular transfusion therapy can help prevent recurring strokes in children at high risk.
- Hydroxyurea.
The first approved drug for the causative treatment of sickle-cell anaemia, hydroxyurea was shown to decrease the number and severity of attacks and shown to possibly increase survival time. This is achieved, in part, by reactivating fetal haemoglobin production in place of the haemoglobin S that causes sickle-cell anaemia. Hydroxyurea had previously been used as a chemotherapy agent, and there is some concern that long-term use may be harmful, but this risk has been shown to be either absent or very small and it is likely that the benefits outweigh the risks. Patients taking the drug needed fewer blood transfusions.
- Dietary cyanate
In the laboratory, cyanate and thiocyanate irreversibly inhibit sickling of red blood cells drawn from sickle cell anemia patients. (How much cyanate in a Cassava meal?)
However, the cyanate would have to be administered to the patient for a lifetime, as each new red blood cell created must be prevented from sickling at the time of creation.
Cyanate also would be expelled via the urea of a patient every cycle of treatment. Also see nicosan(%27globalWrapper%27).style.position%3D%27relative%27%3B., a phytochemical (ethanol/water extract of Piper guineenses seeds, Pterocapus osum stem, Eugenia caryophyllum fruit, and Sorghum bicolor leaves).
- Analgesics
Painful crises. are treated symptomatically with oral and intravenous hydratation and a plan of analgesics progressively stronger: pain management requires in the first time administration of paracetamol at regular intervals until the crisis has settled.
For more severe crises, a subgroup of patients manage on NSAIDs (such as diclofenac or naproxen).
At the last time, patients require opioids and only when the patients can’t control the painful crisis require inpatient management for intravenous.
Diphenhydramine is also an effective agent that is frequently prescribed by doctors in order to help control any itching associated with the use of opioids.
- Folic acid. and penicillin.
Under closer observation by the pediatrician and the management by a hematologist they'll try to remain healthy.
These patients will take a 1 mg dose of folic acid daily for life.
From birth to five years of age, they will also have to take penicillin daily and they have to benefit from routine vaccination for H. influenzae, S. pneumoniae, and Neisseria meningitidis due to the immature immune system.
- Bone marrow transplants
They have proven to be effective in children.
- Gene therapy is under investigation.
- Another treatment being investigated is Senicapoc.