Written by CENA Tiziana, CLEMENTE Emiliano, MORETTI Alessandro
INTRODUCTION
Lysosomal storage disorder (LSDs) (Cartoon) comprise a group of at least 50 distinct genetic diseases, each one resulting from a deficiency of a particular lysosomal protein/activity or, in a few cases, from non-lysosomal activities that are involved in lysosomal biogenesis or protein maturation. Gaucher disease is the most common of the LSDs, followed by Fabry disease. Other diseases in this family include Niemann-Pick disease, Farber diseaser the gangliosidoses (including Tay-Sachs disease), Krabbé disease, and Wolman’s disease.
DEFINITION
Gaucher disease is an autosomal recessive disease characterized by lysosomal accumulation of glucosylceramide (glucocerebroside). Gaucher disease result from defects in the gene encoding the lysosomal hydrolase glucocerebrosidase (acid beta-glucosidase) or rarely, of its activator saposin C. Glucocerebrosidase has the task of transforming the glucocerebroside in sugar (glucose) and fats Ceramides

Structure of a Glucocerebroside
Gaucher disease has traditionally been divided in to the following three clinical subtypes, delineated by the absence or presence of neurologic involvement and its progression:
-Type 1: non neuronopathic form
-Type 2: acute neuronopathic form
-Type 3: chronic neuronopathic form
EPIDEMIOLOGY
The prevalence of Gaucher's disease is low. Types II and III, which have a variable degree of involvement of the neurologic system, have a very rare incidence, and occur in less than 1:100,000 of the population. Type I Gaucher's disease occurs mainly in adults and is the most common lysosomal storage disorder. Experience in the UK suggests over 90 to 95% of patients have predominantly Type I disease, although it is still rare, occurring in about one in 30–40,000 of the population. However, epidemiological studies conducted in the USA and Israel have shown that the incidence of this disorder amongst Ashkenazi Jews is significantly higher than this, with a prevalence of about 1 in 450 in this population (High frequency of the Gaucher disease mutation at nucleotide 1226 among Ashkenazi Jews, 1991), and an estimated carrier frequency of 8,9%. However accurate estimates of the prevalence of this disease have not been available because an unknown number of patients are entirely asymptomatic.
GENETICS
The glucocerebrosidase gene is located on chromosome 1q21 (spans 7 kb encompassing 11 exons) and over 200 mutations associated with Gaucher disease have been identified (Gaucher's disease 2001) In the Ashkenazi Jewish population, the predominant mutation is 1226 A --> G, resulting in an Asp --> Ser substitution at amino acid 370 (N370S). This mutation accounts for ~75% of the mutant alleles in Jewish patients and ~30% in non-Jewish patients. It predisposes to type 1 disease and precludes neurological involvement. Compound heterozygosity with N370S predicts a more severe disease, although there is still absence of neurological symptoms. A frameshift mutation resulting in the insertion of a guanine at nucleotide 84 (84GG) is also common in the Jewish population. The 1448T>C (L444P) mutation is common in the Norbottnian population and the homozygous state has a very high association with the neuronopathic variants of Gaucher disease. The five most common mutations account for ~97% of the alleles in the Jewish population, but ~75% in the non-Jewish.
Acid β-glucosidase exists as a homodimer in human cells. In addition to the enzyme itself, an active hydrolytic complex requires an additional activator protein. The activator of acid β-glucosidase is saposin C, a member of the saposin family of small glycoproteins. The saposins (A, B, C, and D) are all derived from a single precursor, prosaposin. The mature saposins, as well as prosaposin, activate several lysosomal hydrolases involved in the metabolism of various sphingolipids. Prosaposin is proteolytically processed to saposins A, B, C and D, within lysosomes but also exists as an integral membrane protein not destined for lysosomal entry. Uncleaved prosaposin can be found in many biological fluids such as seminal plasma, human milk, and cerebrospinal fluid, where it appears to have a different function.
Saposin C mutations in Gaucher disease patients resulting in lysosomal lipid accumulation, saposin C deficiency, but normal prosaposin processing and sorting, 2010
Specific saposin C deficiency: CNS impairment and acid beta-glucosidase effects in the mouse, 2010
SYMPTOMS
Gaucher disease (GD) encompasses a spectrum of clinical findings from a perinatal-lethal form to an asymptomatic form. However, for the purposes of determining prognosis and management, the classification of GD by clinical subtype is still useful in describing the wide range of clinical findings and broad variability in presentation. Three major clinical types are delineated by the absence (type 1) or presence (types 2 and 3) of primary central nervous system involvement
- Type 1 GD
Bone disease:
Clinical or radiographic evidence of bone disease occurs in 70%-100% of individuals with type 1 GD. Bone disease ranges from asymptomatic osteopenia to focal lytic or sclerotic lesions and osteonecrosis (Skeletal aspects of Gaucher disease: a review, 2002). Bone involvement, which may lead to acute or chronic bone pain, pathologic fractures, and subchondral joint collapse with secondary degenerative arthritis, is often the most debilitating aspect of type 1 GD (Bone and joint complications related to Gaucher disease, 2000). Acute bone pain manifests as 'bone crises' or episodes of deep bone pain that are usually confined to one extremity or joint ("Bone crises in Gaucher disease, 2003":) and are often accompanied by fever and leukocytosis but sterile blood culture. The affected region may be swollen and warm to touch; imaging studies may reveal signal abnormalities consistent with localized edema or hemorrhage; x-rays may show periosteal elevation ('pseudo-osteomyelitis') .
Conventional radiographs (x-rays) may reveal undertubulation (Erhlenmeyer flask configuration) noted in the distal femur and endosteal scalloping as a sign of bone marrow infiltration. MRI reveals the extent of marrow involvement and the presence of fibrosis and/or infarction. In general, marrow infiltration extends from the axial to the appendicular skeleton, and greater involvement is often seen in the lower extremities and proximal sites of an affected bone. The epiphyses are usually spared, except in advanced cases. Bone densitometry studies enable quantitative assessment of the degree of osteopenia.
Although individuals with type 1 GD do not have primary CNS disease, neurologic complications (spinal cord or nerve root compression) may occur secondary to bone disease (e.g., severe osteopenia with vertebral compression; emboli following long bone fracture), or coagulopathy (e.g., hematomyelia) (A neurological symptom survey of patients with type I Gaucher disease, 2003)
Hepatosplenomegaly:
The spleen is enlarged (i.e., 1500-3000 cc in size, compared to 50-200 cc in the average adult) with resultant hypersplenism associated with pancytopenia (i.e., anemia, leukopenia, and thrombocytopenia). Infarction of the spleen can result in acute abdominal pain. Rarely, acute surgical emergencies may arise because of splenic rupture (Life-threatening splenic hemorrhage in two patients with Gaucher disease, 2000)
Liver enlargement is common, although cirrhosis and hepatic failure are rare.
Coagulation abnormalities:
Acquired coagulation factor deficiencies include low-grade disseminated intravascular coagulation and specific inherited coagulation factor deficiencies (e.g., factor XI deficiency among Ashkenazi Jews). An investigation of Egyptian individuals with type 1 GD revealed a wide variety of coagulation factor abnormalities (fibrinogen, factor II, VII, VIII, X, XII) (Coagulation abnormalities in type 1 Gaucher disease in children, 2006). Abnormal platelet aggregation may contribute to bleeding diathesis in the presence of normal platelet counts.
Cytopenias:
Cytopenia is almost universal in untreated GD. Anemia, thrombocytopenia, and leukopenia may be present simultaneously or independently (Survey of hematological aspects of Gaucher disease, 2005). The pattern of cytopenia in GD is dependent on spleen status.
Low platelet count may result from hypersplenism, splenic pooling of platelets, or marrow infiltration or infarction. Immune thrombocytopenia has also been reported and should be excluded in individuals with persistent thrombocytopenia despite GD-specific therapy. Thrombocytopenia may be associated with easy bruising or overt bleeding, particularly with trauma, surgery, or pregnancy. The risk for bleeding may be increased in the presence of clotting abnormalities.
Anemia may result from hypersplenism, hemodilution (e.g., pregnancy), iron deficiency or B12 deficiency and, in advanced disease, decreased erythropoiesis as a result of bone marrow failure from Gaucher cell infiltration or medullary infarction.
Leukopenia is rarely severe enough to require intervention. Deficient neutrophil function has been reported.
Pulmonary involvement:
The following can be observed:
• Interstitial lung disease
• Alveolar/lobar consolidation
• Pulmonary hypertension; well documented in individuals with liver disease and presumably the result of inability to detoxify gut-derived factors, which somehow adversely affect the pulmonary endothelium with resultant pulmonary hypertension. Pulmonary hypertension can also occur in individuals with GD without liver disease . (Pulmonary hypertension in type 1 Gaucher's disease: genetic and epigenetic determinants of phenotype and response to therapy, 2002) Dyspnea and cyanosis with digital clubbing attributed to hepatopulmonary syndrome have been described in individuals with liver dysfunction, often caused by an intercurrent disease (e.g., viral hepatitis).
Pregnancy and childbirth:
Except in women with significant pulmonary hypertension, pregnancy is not contraindicated in GD.
Pregnancy may affect the course of GD both by exacerbating preexisting symptoms and by triggering new features such as bone pain. Women with severe thrombocytopenia and/or clotting abnormalities may have an increased risk of bleeding around the time of delivery.
In some women the diagnosis of GD is first made in pregnancy because of exacerbation of hematologic features.
Pregnancies in Gaucher disease: a 5-year study, 2004
Malignancy:
Epidemiologic studies have suggested elevated risk of certain malignancies in GD including the following:
• Multiple myeloma
• Hepatocellular carcinoma
• Non-Hodgkins lymphoma, malignant melanoma, and pancreatic cancer
Increased incidence of cancer in adult Gaucher disease in Western Europe, 2006
Gaucher disease and cancer incidence: a study from the Gaucher Registry, 2005
Increased risk of cancer in patients with Gaucher disease, 1993
Other reports have failed to find these associations.
Immunologic abnormalities:
Children or adults may have polyclonal gammopathy. Affected individuals also exhibit altered cellular immune profiles with increased peripheral blood NKT lymphocytes and reduced numbers of functionally normal dendritic cells.
(Hyperimmunoglobulinemia in pediatric-onset type 1 Gaucher disease and effects of enzyme replacement therapy, 2007)
Metabolic abnormalities:
GD is associated with metabolic abnormalities including high resting energy expenditures (possibly the result of elevated cytokine levels) and low circulating adiponectin and peripheral insulin. The hypermetabolic state is not associated with altered thyroid hormone resistance
Hypermetabolism in Gaucher disease type I is not associated with altered thyroid hormone levels, 2007
Psychological complications:
Persons with GD exhibit moderate to severe psychological complications including somatic concerns and depressed mood.
Psychological complications of patients with Gaucher disease, 2006
- Type 2 GD / Type 3 GD (Primary Neurologic Disease)
Neurologic disease:
Previously, affected individuals were classified into type 2 or type 3 GD based on the age of onset of neurologic signs and symptoms and the rate of disease progression. Children with onset before age two years with a rapidly progressive course, limited psychomotor development, and death by age two to four years were classified as having type 2 GD. Individuals with type 3 GD may have onset before age two years but often have a more slowly progressive course, with life span extending into the third or fourth decade in some cases. However, these distinctions are not absolute and it is increasingly recognized that neuropathic GD represents a phenotypic continuum, ranging from abnormalities of horizontal ocular saccades at the mild end to hydrops fetalis at the severe end.
Bulbar signs include stridor, squint, and swallowing difficulty.
Pyramidal signs include opisthotonus, head retroflexion, spasticity, and trismus.
Oculomotor apraxia, saccadic initiation failure, and opticokinetic nystagmus are common .
Oculomotor involvement may be found as an isolated sign of neurologic disease in individuals with a chronic progressive course and severe systemic involvement (e.g., massive hepatosplenomegaly).
Generalized tonic-clonic seizures and progressive myoclonic epilepsy have been observed in some individuals.
Dementia and ataxia have been observed in the later stages of chronic neurologic disease.
Ocular motor abnormalities in Gaucher disease, 1999
Phenotypic continuum in neuronopathic Gaucher disease: an intermediate phenotype between type 2 and type 3, 2003
Myoclonus from selective dentate nucleus degeneration in type 3 Gaucher disease, 2003
Perinatal-lethal form:
The perinatal-lethal form is associated with hepatosplenomegaly, pancytopenia, and microscopic skin changes (i.e., abnormalities in the stratum corneum attributed to altered glucosylceramide-to-ceramide ratio) and may present clinically with ichthyosiform or collodion skin abnormalities or as nonimmune hydrops fetalis . Arthrogryposis and distinctive facial features are seen in 35%-43%
Another rare severe variant of GD is associated with hydrocephalus, corneal opacities, deformed toes, gastroesophageal reflux, and fibrous thickening of splenic and hepatic capsules.
Glucocerebrosidase gene mutations in patients with type 2 Gaucher disease, 2000
A new variant neuropathic type of Gaucher's disease characterized by hydrocephalus, corneal opacities, deformed toes, and fibrous thickening of spleen and liver capsules, 2001
Cardiovascular form:
Individuals homozygous for the D409H allele present with an atypical phenotype dominated by cardiovascular disease with calcification of the mitral and aortic valves. Additional findings include mild splenomegaly, corneal opacities, and supranuclear ophthalmoplegia .
Gaucher disease with oculomotor apraxia and cardiovascular calcification (Gaucher type IIIC), 2000
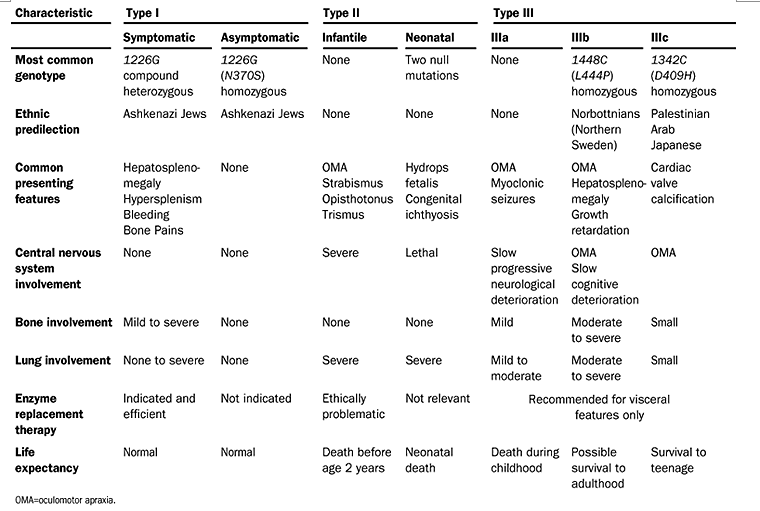
Elstein et al. Lancet 2001;358:324-327.
DIAGNOSIS
You can reach the diagnosis using the following methods:
Enzyme analysis:
The most effective and reliable diagnostic method is the measurement of glucocerebrosidase activity in leukocytes from peripheral blood or in cultures of fibroblasts. In general, patients with Gaucher disease have enzyme activity less than 30% of normal activity. In some patients, the correct diagnosis can be suspected initially from a bone marrow aspirate who showing Gaucher cells. The enzymatic test that uses a spot of dried blood on filter cards (DBS) can be used for the diagnosis of the disease.(Gaucher and Niemann-Pick diseases--enzymatic diagnosis in dried blood spots on filter paper: retrospective diagnoses in newborn-screening cards, 2002)
Genetic mutation analysis:
Primers for the polymerase chain reaction were designed to discriminate between mutant and wild-type alleles of glucocerebrosidase and to allow separation from products of the related pseudogene. The nucleotide 1226 mutation (asparagine 370→ serine) and 84GG (an insertional frameshift mutation) were found exclusively in five patients of Ashkenazi Jewish descent (7 and 2 of the 10 disease alleles, respectively). Two point mutations, at nucleotides 1448 (leucine 444→proline) and 1504 (arginine 463→cysteine), were found in 4 and 3 alleles, respectively; they were associated with rapidly progressive disease and neurological involvement in non-Jewish patients. The ARMS procedure for direct detection of common mutations in glucocerebrosidase will facilitate genetic counselling and screening programmes for individuals at risk of Gaucher's disease.
(Genetic diagnosis of Gaucher's disease, 1992(92)90928-V/abstract)
(Mutation analysis of 28 Gaucher disease patients: the Australasian experience, 1994)
Prenatal testing:
For pregnant women who are carriers of the Gaucher's gene, doctors may recommend prenatal testing for genetic mutations that can determine whether the fetus is at risk of Gaucher's disease. Tests that evaluate cells in the amniotic fluid (amniocentesis) or evaluate tissue from the placenta (chorionic villus sampling) can detect all types of Gaucher's in the fetus.
Prenatal molecular diagnosis of Gaucher disease, 1995
TRACKING THE PROGRESSION:
If you receive a diagnosis of Gaucher's disease, your doctor may recommend periodic tests to track its progression. These may include imaging tests such as:
• Dual energy X-ray absorptiometry (DXA), which uses low-level X-rays to measure bone density, including changes over time.
• Magnetic resonance imaging (MRI) scans, which use magnetic fields and radio waves to create images in order to highlight conditions of hepatosplenomegaly.
Gaucher disease: new molecular approaches to diagnosis and treatment, 1992
PATHOGENESIS
Enzymatic degradation of cell constituents takes place primarily in secondary lysosomes. Secondary lysosomes, also known as phagolysosomes, form when primary lysosomes fuse with phagocytic vacuoles containing the ingested material.
Gaucher disease is due to a hereditary deficiency in the activity of glucocerebrosidase, one of the lysosomal enzymes required for glycolipid degradation.
Macrophages are the primary cells affected, and include all cells of the mononuclear phagocyte system, especially Kupffer cells in the liver, osteoclasts in bone, microglia and CSF macrophages in the central nervous system, alveolar macrophages in the lungs, and histiocytes in the spleen, lymph nodes, bone marrow, gastrointestinal and genitourinary tracts, as well as the peritoneum. They have been described in almost every organ in the body.
This illustration demonstrating how cytoplasmic material accumulates in Gaucher disease :
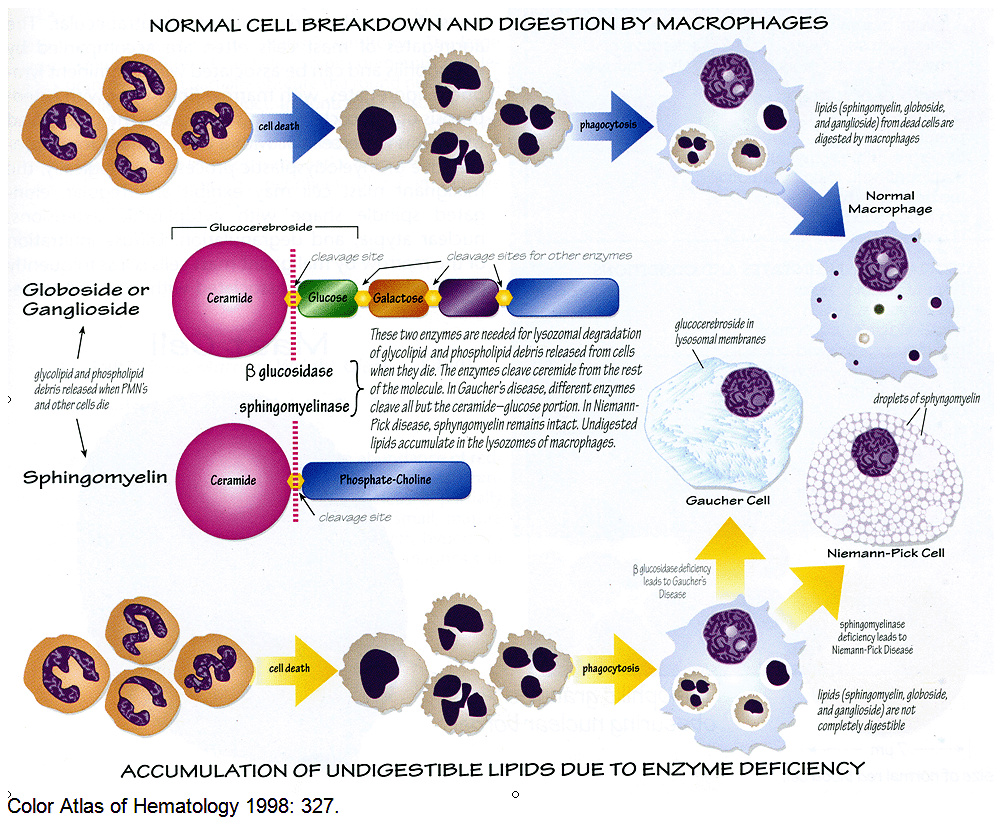
Although the liver and spleen may increase greatly in size, the amount of pathological lipid stored in the affected macrophages (Gaucher cells) accounts for less than 2% of the additional tissue mass. It is therefore clear that an inflammatory response occurs in affected individuals and that the clinical phenotype is due to an effect of macrophage storage beyond the physical presence of the Gaucher cells. Factors released by Gaucher cells, including pro-inflammatory cytokines and perhaps cathepsins, provide a mechanistic link between lysosomal storage and the diverse clinical manifestations of Gaucher disease.
Pro-inflammatory cytokines and the pathogenesis of Gaucher's disease: increased release of interleukin-6 and interleukin-10, 1997
Impaired IL-10 transcription and release in animal models of Gaucher disease macrophages, 2009
This prevailing macrophage-centric view, however, does not explain emerging aspects of the disease, including malignancy, autoimmune disease, Parkinson disease, and osteoporosis: a recent study showed that widespread dysfunction is not only of macrophages, but also of thymic T cells, dendritic cells, and osteoblasts (Glucocerebrosidase gene-deficient mouse recapitulates Gaucher disease displaying cellular and molecular dysregulation beyond the macrophage 2010 ) .
Interaction between parkin and mutant glucocerebrosidase variants: a possible link between Parkinson disease and Gaucher disease,2010
Gaucher disease ascertained through a Parkinson's center: imaging and clinical characterization,2010
The risk of Parkinson's disease in type 1 Gaucher disease
THERAPY
In the past, patient care and therapy for Type 1 Gaucher disease was only aimed to managing or relieving symptoms. Treatments included various pain reduction therapies, blood transfusions, orthopedic surgery for bones and joints, and possible splenectomy. Although many of these measures still have a place in the management of Type 1 Gaucher disease, the focus of disease management shifted in the early 1990s with the advent of disease-specific therapy. There are two major approaches to disease-specific therapies for Type 1 Gaucher disease: enzyme replacement therapy and substrate reduction therapy. These therapeutic approaches are also used in other lysosomal storage diseases and the descriptions below are intended to give you a general overview of these approaches.
Enzyme replacement therapy (ERT):
The goal of ERT is to provide the right amount of enzyme to allow for the proper disposal of accumulated substances. Thus, enzyme replacement therapy works by supplementing or replacing the Gaucher patient’s missing or deficient enzyme. With ERT, smaller components of waste can be removed from cells by natural processes. However, ERT does not currently address conditions or symptoms related to the central nervous system of Types 2 and 3 Gaucher disease.
The two recombinant glucosylceramidase enzyme preparations currently available commercially are based on the human gene sequence and are distinguished according to the cell type involved in their production: imiglucerase (Cerezyme®) generated in Chinese hamster ovary cells; velalglucerase alfa (VPRIV®) from human cell line.
After prolonged treatment, ERT reduces the rate of bone loss in a dose-dependent manner ,improves bone pain, and reduces bone crises in individual with tipy 1 gaucher diasease.
The effect of enzyme replacement therapy on bone crisis and bone pain in patients with type 1 Gaucher disease,2007
Effectiveness of enzyme replacement therapy in 1028 patients with type 1 Gaucher disease after 2 to 5 years of treatment: a report from the Gaucher Registry, 2002
Substrate reduction therapy (SRT):
Substrate reduction therapy with inhibitors of glucocerebroside formation has been approved for patients in whom enzyme replacement therapy is "unsuitable" or "not a therapeutic option."
Miglustat (N-butyldeoxynojirimycin) is the only substrate reduction intervention thus far licensed, and causes a reduction in hepatosplenomegaly, but a slower response of hematological parameters.
Miglustat (Zavesca) in type 1 Gaucher disease: 5-year results of a post-authorisation safety surveillance programme, 2005
Gaucher disease and the clinical experience with substrate reduction therapy,2003
Psychological Care
It's also important to consider the mental and emotional impact that Gaucher disease can place on patients and their families. Professional counseling can help patients better manage the difficulties of their disease and the lifestyle changes that may be required.
Future treatments may include gene therapy and intervention with chemical chaperones. The latter have demonstrated some capacity to stabilize or reactivate improperly folded glucocerebrosidase in vitro.
Enzyme Replacement in Gaucher Disease, 2004
THERAPIES UNDER INVESTIGATION
Enzyme replacement therapy (ERT): An alternative recombinant enzyme formulation generated from transduced plant (carrot) cells (taliglucerase alfa), is currently in clinical trials . This enzyme preparation also has exposed mannose residues involved in intracellular uptake.
Production of glucocerebrosidase with terminal mannose glycans for enzyme replacement therapy of Gaucher's disease using a plant cell system, 2007
Substrate reduction therapy (SRT): Eliglustat, an alternative inhibitor of glucosylceramide synthetase currently under clinical evaluation, has shown in a phase I/II study to be effective in reversing the hematologic, visceral, and skeletal manifestations of GD .Eliglustat is metabolized by CYP345, which implies potential for drug interactions and thus requires monitoring not currently undertaken with ERT.
Improvement in hematological, visceral, and skeletal manifestations of Gaucher disease type 1 with oral eliglustat tartrate (Genz-112638) treatment: 2-year results of a phase 2 study, 2010