The Keshan Disease
Definition
Keshan disease is a congestive cardiomyopathy probably caused by dietary deficiency of selenium, named after Keshan, where symptoms were first noted. These symptoms were later found prevalent in young women and children in a wide belt extending from northeast to southwest China, a region with a selenium-deficient soil. Others Authors had linked it to a Coxsackie B virus infection. However, the exact etiology has not been clarified up to now.
Wikipedia
PubMed
Epidemiology

Keshan’s disease Area.

Selenium-deficit Area.
Epidemiological surveys have shown that Keshan disease occurs in large areas in 14 provinces in China, mainly in the countryside. The largest prevalence age rates are boys under 15 years old and women of childbearing age. However, the incidence of Keshan disease varies according to area, seasons and population. The prevalence of Keshan disease was found to vary from year to year. In the past, there have been frequent outbreaks of the disease. For example, the incidence of Keshan disease in 1955 was 32.7/100.000 in Heilongjiang Province verus a common annual incidence of 1.73/100.000. The 99% of the patients were peasants whose diet consisted of home-grown grain. The incidence of Keshan disease has sharply decreased after 1980 along with the steady raise of living standard and realization of preventive measures.
Springer Link
PubMed
Etiology
Selenium supplementation protects people from developing Keshan disease but cannot reverse heart muscle damage once it occurs. Despite the strong evidence that selenium deficiency is a fundamental factor in the etiology of Keshan’s disease, the seasonal and annual variation in its occurrence suggests that an infectious agent is involved in addition to selenium deficiency. Coxsackievirus is one of virus type that has been isolated from Keshan patients, and studies in selenium-deficient mice show that this virus is capable of causing an inflammation of the heart called myocarditis.
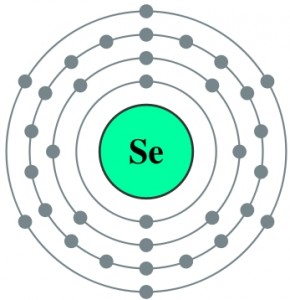
The discovery that Keshan disease likely has a dual etiology that involves both a nutritional deficiency of the essential trace mineral selenium (Se) as well as an infection with an enterovirus provided the impetus for additional studies of relationships between nutrition and viral infection. An amyocarditic strain of coxsackievirus B3, CVB3, converted to virulence when it was inoculated into Se-deficient mice. This conversion was accompanied by six nucleotidie changes in the genetic structure of the virus so that its genome closely resembled that of other known virulent CVB3 strains. Similar alterations in virulence and genomic composition of CVB3 could be observed in mice fed normal diets but genetically deprived of the antioxidant selenoenzyme glutathione peroxidase (knockout mice).
The role of nutrition in infectious disease has long been associated with changes in the immune response of the nutritionally deficient host. It has been shown in a number of studies that nutritionally deficient humans or animals are more susceptible to a wide variety of infections. This increase in susceptibility is thought to be the result of an impaired host immune response due to a deficient diet.
However, recent studies have demonstrated that not only is the host immune response affected by the deficient diet, but the viral pathogen itself can also be altered. Dietary deficiencies that lead to oxidative stress in the host [e.g., selenium (Se) deficiency] can alter a viral genome such that a normally benign or mildly pathogenic virus becomes highly virulent in the deficient, oxidatively stressed host. Once the viral mutations occur, even hosts with normal nutriture can be affected by the newly pathogenic strain.
The Journal of nutrition
Pathogenetic hypothesis
To understand the role of Selenium in Keshan disease we have to remind the role of Selenium in selenium-dependent enzymes, also known as selenoproteins, and in particular about Glutathione Peroxidases: during selenoprotein synthesis, selenocysteine is incorporated into a very specific location in the amino acid sequence in order to form a functional protein. Five selenium-containing glutathione peroxidases (GPx) have been identified: cellular or classical GPx, plasma or extracellular GPx, phospholipid hydroperoxide GPx, gastrointestinal GPx, and olfactory GPx. Although each GPx is a distinct selenoprotein, they are all antioxidant enzymes that reduce potentially damaging reactive oxygen species (ROS), such as hydrogen peroxide and lipid hydroperoxides, to harmless products like water and alcohols by coupling their reduction with the oxidation of glutathione.
One molecule of hydrogen peroxide is reduced to two molecules of water, while two molecules of glutathione (GSH) are oxidized in a reaction catalyzed by the selenoenzyme, glutathione peroxidase. Oxidized glutathione may be reduced by the flavin adenine dinucleotide (FAD)-dependent enzyme, glutathione reductase.

The possible relationship between a deficiency in Se and infection with coxsackievirus was explored further in many laboratories: weanling mice were fed a diet deficient in Se for 4 weeks before intraperitoneal inoculation with an amyocarditic strain of coxsackievirus B3 (CVB3). In normal mice, this virus does not induce cardiac inflammation, although it will replicate in heart cells.
Se is an essential component of the antioxidant enzyme GPx. Serum samples from the mice were tested for the presence of GPx activity as a biomarker for Se status. Mice that were fed a diet deficient in Se had a fivefold decrease in GPx activity as compared with mice fed a diet adequate in Se. As expected, infected mice that were fed a diet adequate in Se did not develop myocarditis. However, infected mice that were fed a diet deficient in Se developed myocarditis. In addition, heart viral titers were elevated in the Se-deficient mice as compared with the Se-adequate mice.
To determine whether the Se status affected the immune response of the infected mice, serum neutralizing-antibody titers and spleen cell proliferative responses were determined. Levels of neutralizing antibody in the Se-deficient mice were equivalent to levels found in the Se-adequate mice, although the ability of spleen cells in vitro to proliferate in response to either mitogen or CVB3 antigen was severely impaired in the Se-deficient mice. In addition, mRNA for the chemokine macrophage chemotactic protein-1 and interferon-γ were overexpressed in the Se-deficient mice. Thus, it appeared that the deficiency in Se had altered the host immune response to infection with CVB3.
Because Se is involved in the functioning of enzymes other than GPx, we wanted to determine whether the virulence of CVB3 in the Se-deficient mice was due to a deficiency in GPx activity. To establish a relationship between GPx activity and Se deficiency, we used GPx-1–knockout mice. All Se-containing enzymes except GPx-1 are fully functioning in these knockout animals. GPx-1–knockout mice were infected with CVB3 and killed at various times postinfection. Myocarditis developed in >50% of the knockout mice, which suggests that antioxidant protection is important for protection against CVB3-induced myocarditis. Interestingly, in contrast to the immune response of the Se-deficient mice, the GPx-1–knockout mice had reduced serum neutralizing-antibody production, although their spleen cells were able to respond to both mitogen and antigen. The ability of the normally amyocarditic CVB3 to induce myocarditis in Se-deficient and GPx-1–knockout mice may have been due to effects on the immune system that left the mice vulnerable to the virus. Alternatively, the virus itself may have been altered as a consequence of replicating in an oxidatively stressed host. To determine which of these possibilities is correct, a viral passage experiment was performed.
Se-deficient mice were infected with CVB3. At 7 days postinfection, mice were killed and the virus was isolated from their hearts. This virus was then passed back into Se-adequate mice. If the pathogenicity of the virus was due to host factors alone, then the Se-adequate mice should not develop myocarditis. However, the Se-adequate mice did develop myocarditis, which suggests that the virus itself had changed. Virus passed from Se-adequate to Se-adequate mice did not result in pathology, which demonstrates that viral passage alone did not alter the virus.
To confirm that the phenotype change of the virus was due to a change in viral genotype, we sequenced virus that was isolated from Se-adequate and Se-deficient mice and compared those sequences to the sequence of the input strain. We found six nucleotide changes between the original CVB3 strain and the virus isolated from Se-deficient mice: Nt 234: C → T, Nt 788: G → A, Nt 2271: A → T, Nt 2438: G → C, Nt 3324: C → T and Nt 7334: C → T. No changes were found in virus that was isolated from Se-adequate mice.
Phosphorylation of JNK1/2 and p38 MAPK increase during active replication of CVB3 and their phosphorylation was independent of CVB3-induced caspase activation or production of reactive oxygen species. The roles of these kinases in CVB3 infection were further evaluated using specific inhibitors. JNK1/2 inhibitors reduced CVB3-induced phosphorylation of activating transcription factor 2, and the p38 MAPK inhibitor reduced CVB3-induced phosphorylation of heat shock protein 27. Although inhibition of these kinases by specific inhibitors did not affect CVB3 viral protein synthesis, inhibition of p38 MAPK but not of JNK1/2 resulted in significant reduction of viral progeny release, suppression of CVB3-induced cell death, and blockage of CVB3-induced caspase-3 activation in infected cells.
A strong immunopathologic component in CVB3-induced disease and implicate both CD41and CD8 T cells. Compared with immunocompetent animals, mice lacking CD4 T cells (CD4ko) were more resistant to virus challenge, and mice lacking CD8 T cells (b2Mko and in vivo-depleted CD4ko) showed enhanced survival and a reduced incidence of the later myocarditis. Nevertheless, the picture is complex, since removal of the CD4 component, while protecting against early death, greatly magnified the severity of myocarditis, and removal of the CD8 cells from CD4ko mice, although protecting against early death and later myocarditis, led to markedly increased virus titers in the heart. These data underscore the complex balance between the costs and benefits of effective antiviral immune response
This is the first report of a specific host nutritional deficiency driving changes in a viral genome and changing a normally avirulent pathogen into a virulent one. Once these changes occur, even hosts with normal nutritional status are vulnerable to the newly virulent virus. However the mechanisms responsible for the changes is currently under investigation.
Similar to virus that replicated in a Se-deficient host, virus isolated from GPx-1–knockout mice that developed myocarditis also had viral genome changes. Virus isolated from GPx-1–knockout hearts that developed myocarditis had the same six mutations as were found in virus isolated from Se-deficient mice. In addition, another nucleotide change occurred: Nt 2690: G → A. The changes in the genome were associated with the increased virulence of the virus, because viruses isolated from GPx-1–knockout mice that did not develop myocarditis were identical to the input strain.
Thus, both a deficiency in Se as well as a specific deficiency in GPx-1 activity lead to viral genome changes, which suggests that increased oxidative stress of the host can affect a viral genome.
The Journal of nutrition
Pubmed Pubmed Pubmed
Linus Pauling Institute
Pathology
Multifocal necrosis and fibrous replacement of the myocardium characterized the histopathological features and myocytolysis was present in most cases. The onset of the disease could be Acute, Subacute or Chronic. Ultrastructural observations on twelve postmortem cases revealed remarkable changes in many organelles of which mitochondria appeared to be most important in the development of myocardial lesions. Probably Keshan disease is a form of “mitochondrial cardiomyopathy”.
Pubmed
Science in China Series C: Life Sciences 2006 Vol.49 No.6 513 518 – Keshan disease and mitochondrial cardiomyopathy
Signs and Symptoms
The most important sign is enlarged heart. Other symptoms are higher rates of cancer, cardiovascular disease, hypertension, and strokes. An individual can also have eczema, psoriasis, arthritis, cataracts, alcoholism, and infections.
Initially there may be symptoms like diarrhea, vomiting, fever and body pains.
The initial signs include heart inflammation (increase of Ves, PCR, TnI, Cpk), Arrhythmia, Cardiac insufficiency.
Advanced stage is characterized by Pulmonary Edema, Low Blood Pressure and Ejection Fraction, Heart Failure and death.
Health Care Nutrition
Treatment
Treatment for this condition is by supplementation of Se in diet. Many initial intervention studies showed the prophylactic effect of the inorganic Sodium Selenite. If treated early the effects of this aliment can be reversed. However damage to heart muscle cannot be reversed and the supplements can only stop further progress of the condition. If necessary and if possible, surgery is performed to correct the heart condition.
Individuals in China with the disease, treat it with a herb called Astragalus, which accumulates selenium from the soil.
Matteo Bianco, Simone Galotti