Male infertility refers to the inability of a male to achieve a pregnancy in a fertile female. In humans it accounts for 40-50% of infertility. Male infertility is commonly due to deficiencies in the semen and semen quality is used as a surrogate measure of male fecundity
EPIDEMIOLOGY
Infertility is a major health problem affecting 10-15% of couples seeking to have children, and a male factor can be identified in about half of these cases.
Infertility in Men: Recent Advances and Continuing Controversies
A significant proportion of infertile males are affected either by oligozoospermia (reduced sperm production) or azoospermia (lack of any sperm in the ejaculate). Such alterations in sperm production may be related, in turn, to different underlying testicular histological pathologies, ranging from the complete absence of germ cells (Sertoli cell-only syndrome SCO) to hypospermatogenesis and maturation arrest. The alteration of spermatogenesis can be the consequence of many causes, such as sistemic disease, cryptorchidism, endocrinological disorders, obstruction/absence seminal patwhays, or infections.
Prevalence:
- Infertility affects approximately 15 percent of couples of reproductive age. Male-factor infertility is a factor in one-half of these cases.
- Approximately 15–20 percent of infertile men are azoospermic.
SYMPTOMS
Male infertility is characterised by four main categories:
- Azoospermia :complete absence of sperm cells in semen and it can be classified into three types:
- Pre-testicular (in about 2% of azoospermia) : is characterized by inadequate stimulation of a normal testicles and normal genital tract. Typically FSH levels are low commensurate with inadequate stimulation of the testes to produce sperm. Example include hypopituitarism, hyperprolactinemia, chemotherapy
- Testicular (in about 49-93% of azoospermia): in this situation the testes are abnormal atrophic, or absent, and sperm production severly disturbed to absent. FSH level tends to be elevated. Testicular failure includes absence of failure production as well as low production and maturation arrest during the process of spermatogenesis. Example includes Klinefelter syndrome, cryptorchidism or Sertoli cell-only syndrome as well as acquired conditions by infection (orchitis), surgery (trauma, caner), radiation, o other causes.
- Post-testicular_ (in about 7-51% of azoospermic men): in this situation sperm are produced but not ejaculated. The main cause is physical obstruction (obstructive azoospermia) of the post-testicular genital tracts. The most common reason is vasectomy, other obstructions can be congenital (agenesis of the vas deferens as senn in certain case of fcystic fibrosis) or acquired, such as ejaculatory duct obstruction for instance by infection.
- Oligozoospermia :few spermatozoa in semen. Many conditions listed before may also cause various degrees of oligospermia rather than azoospermia.
GENETIC FACTORS
Genetic factors (more than 2,300 genes may play a role in male infertility [Schultz et al.2003] ) can cause prestesticular, testicular, and post-testicular azoospermia (or oligospermia) and include the following situations:
- Mutations in the CFTR gene (cystic fibrosis transmembrane conductance regulator),
CHROMOSMAL ABNORMALITIES
The great majority of observed chromosomal abnormalities are numerical or structural involving sex chromosomes [Yatsenko, et al. 2010]
Infertile men have a major risk to have abnormalities on karyotyping (1.3 to 13.1%) compared to fertile male population (about 0.85%).
The main chromosomal abnomarlities in infertile men are:
- Numerical abnormalities
- 47 XXY Klinefelter's syndrome: 85% of Azoospermia and 15% of severe oligospermia (incidence in normal population: 1:500-1:1000),
- 47 XYY's syndrome: normally or occasionally infertile (incidence in normal population: 1:500 ),
- Structural abnormalities
- Robertsonian transolaction: from normospermia to azoospermia (incidence in normal population: 1:1000),
- Reciprocal translocation: from normospermia to azoospermia (incidence in normal population: 1:500).
Genetic counselling is indicated for men with genetic causes of azoospermia. In terms of reproduction, it needs to be considered if the genetic defect could be transmitted to the offspring.
In humans Klinefelter's Syndrome (KS) 47, XXY is the most common sex chromosome disorder in males. KS affects approximately 1:500-1000 males and the 10% of the azoospermic males presents this patology.
KS is a condition in which human males have an extra X chromosome.
The principal effects are development of small testicles and reduces fertility (azoospermia or severe oligozoospermia). A variety of other physical and behavioral differences and problems are common, though severity varies and many boys and men with the condition have few detectable symptoms ( The most KS's men have the only infertility as symptom).
MUTATIONS IN THE CFTR GENE
Cystic fibrosis transmembrane conductance regulator (CFTR) is a protein that in humans is encoded by CFTR gene situated on the long arm of chromosome 7.
Mutation of the CFTR gene affect functioning of the chloride ion channels in the epithelial cell membranes, leading to cystic fibrosis and congenital absence of the vas deferens (CAVD).
Cystic fibrosis is a common autosomal recessive genetic disease wich affects the entire body, causing progressive disability and often early death.
About 97% of men with cystic fibrosis are infertile due to congenital absence of the vas deferens. They produce normal sperm but are missing the tube (vas deferens), which connects the testes to the ejaculatory ducts of the penis (obstructive azoospermia). This men are infertile but not sterile and can have children with assisted reproductive techniques (testicular sperm extraction: TESE).

Y CHROMOSOME MICRODELETIONS (YCM)
YCM are the second cause of infertility after the KS and are estimated to occurs in one in 2000-3000 males.
Y chromosome microdeletion (YCM) is a family of genetic disorders caused by missing genes in the Y chromosome. Men with YCM exhibit no symptoms and lead normal lives.
However, YCM is presents in a significant number of men with reduced fertility.
The association between male infertility and absence of genes that map to the long arm of Y chromosome was established in 1976 [Tiepolo and Zuffardi 1976] when 0.5% of idiopathic sub infertile individuals were found to carry cytogenetically visible deletions of the Yq11. The authors termed the missing locus ‘azoospermia factor (AZF)’. Several studies using both karyotyping and molecular techniques confirmed the association between deletions involving the Yq11 region and male infertility [Andersson et al. 1988; Chandley et al. 1989]. The use of molecular markers such as Y-chromosome locus-specific probes enabled the detection of small non-overlapping interstitial deletions culminating with the molecular definition of three AZF regions at Yq11 (AZFa, AZFb, and AZFc) that each contain numerous genes involved in spermatogenesis [Ma et al. 1992; Reijo et al. 1995; Vogt et al. 1992; 1996].
These spermatogenesis loci are termed AZFa, AZFb, and AZFc from proximal to distal Yq. Furthermore, a fourth region (AZFd) has been proposed between AZFb and AZFc, but this finding must be confirmed.
According to Vogt et al.:
- AZFa is located at the proximal portion of deletion interval 5 (subinterval 5C),
- AZFb spans from the distal portion of deletion interval 5 to the proximal end of deletion interval 6 (subinterval 5O–6B),
- AZFc is located at the distal part of deletion interval 6 (subintervals 6C–6E).
Several genes located in AZF regions are expressed in the testis and could therefore be viewed as “AZF candidate genes.” However, based on studies of infertile patients, only a few genes can actually be considered responsible for the AZF phenotype.
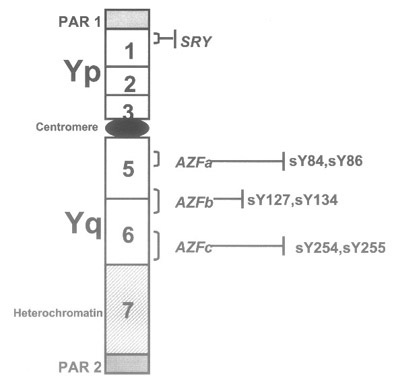
Figure: schematic representation of the Y chromosome. Outside the PARs (pseudoautosomal regions) is the nonrecombining region of the Y chromosome (NRY). This part consists of several repetitive sequences that may be homologs to regions on the X chromosome or Y-specific. The Yp and the proximal part of Yq consist of euchromatin, while the distal part of the long arm is made of heterochromatin.
Therefore, the Y chromosome long arm may be cytogenetically divided in an euchromatic
proximal region (Yq11, subdivided into Yq11.1, 11.21, 11.22, and 11.23) and a heterochromatic distal region (Yq12), whereas the euchromatic short arm is called Yp11.
YCM's genetic:
- Inheritance is Y-linked.
- Y chromosome microdeletions typically occur de novo. Rarely, men carrying a microdeletion may be fertile and father infertile sons.
- Penetrance is near 100 percent in affected males.
- Microdeletions of the azoospermia factor (AZF) regions on the q arm of the Y chromosome are present in 5–10 percent of males with non-obstructive azoospermia or severe oligospermia.
Microdeletions of the AZF regions have different frequency and each shown a diverse clinical phenotype (different spermatogenesi's abnormalities.)
Relative prevalence of deletions in AZFa,b,c region in infertile men:
- AZFa: 5%
- AZFb: 10%
- AZFc: 70%
- AZFa+b: 1.5%
- AZFb+c: 8.3%
- AZFa +b+c: 4%

AZFa
- 5% :rare.
- Complete absence of germ cells.
- SCO: Sertoli-cell-only syndrome
The AZFa region is long about 1.100 KB and the deletions in this part are rare.
AZFa microdeltions represent only the 5% of the all deletions but they are associated to a severe spermatogenic impairment.
The data suggest that more than one gene may be responsible for the AZFa phenotype. This region contains two main genes: USP9Y and DBY
The first gene identified in AZFa and subsequently shown to be absent in infertile patients was USP9Y (ubiquitin specific protease 9, Y chromosome).
USP9Y seems to function as a C-terminal ubiquitin hydrolase and it is ubiquitously expressed in a wide range of tissues, rather than testis specific.
USP9Y occupies less than half of the AZFa interval, while the majority of infertile males carrying AZFa deletions show the absence of this entire interval. These findings suggested that other gene(s) in this region may be responsible, either singly or in combination with USP9Y, for the spermatogenic disruption observed in AZFa-deleted patients.
Initial studies on patients with deletions clearly limited to AZFa suggested that deficiency of USP9Y cause male infertility and that the additional loss of DBY may make the phenotype worse. It is more frequently deleted than USP9Y , and it shows a testis-specific transcript in addition to ubiquitous transcripts.
However, its specific function in male germ cell development is still unknown.
AZF b
- 16%
- Azoospermia with SCO
- Spermatogenic arrest.
Microdeletions can remove AZFb alone, parts of AZFb, or also include flanking regions (AZFc). Two genes have been mapped : EIF1AY (translation-initiation factor 1A, Y isoform) and RBMY (RNA binding motif on the Y). EIF1AY possesses abundant testis-specific transcripts in addition to ubiquitous transcripts, suggesting that this gene may contribute to the AZFb phenotype but it is not considered an AZFb-candidate gene.
RBMY was the first among the AZF candidate genes to be identified.
The RBMY proteins consist of a single RNA-binding domain of the RRM (RNA recognition motif) type at the N terminus and an auxiliary C-terminal domain containing four 37-amino acid (aa) repeats.
Consistent with a role in spermatogenesis, the RBMY genes are expressed only in the germline in the testis (spermatogonia, spermatocytes, and round spermatids). The actual function of RBMY in male germ cell development is not clear; it is a nuclear protein with dynamic modulations in its spatial location in the various spermatogenic cells, suggesting that it possesses different functions related to pre-mRNA splicing.
RBMY is considered the major AZFb candidate gene given its testis specificity, its absence in a fraction of infertile patients, and its homology with the mouse Rbm, deletion of which causes male sterility. However, the multicopy nature of this gene has complicated attempts to prove its role in human spermatogenesis, as detrimental mutations in patients have not yet been identified.
AZFc
- 60%
- Variable phenotype ranging from mild oligospermia to azoospermia and SCO.
One gene has been mapped in this regions and it was named DAZ (deleted in azoospermia). This gene is a member of a multigene family with more than one copy on the Y chromosome, clustered in the AZFc region.
The structure of the DAZ gene is somewhat similar to that of RMBY . DAZ encodes a protein with a single RNAbinding domain at the N terminus and a C-terminal domain containing an internally repeated sequence of 24 aa, the so called DAZ repeats. The DAZ transcription unit appears to contain at least 16 exons and to span about 42 kb. Exon 1 consists of the initiator codon, exons 2–5 encode the RNA-binding domain, and each of exons 7a–7 g encodes a single DAZ repeat.
Like RBMY, DAZ is transcribed and translated into proteins only in male germ cells.
DAZ is found on the Y chromosome only in humans, although DAZ is not the only gene present in the distal Yq interval 6, its high prevalence of deletions in
infertile men makes it the major AZFc candidate.
This possibility is further strengthened by the high homology of DAZ with a Drosophila male infertility gene, mutation of which causes spermatogenic arrest. Furthermore, more recent proof of the spermatogenic role of the DAZ gene product arises from the observation that a human DAZ transgene is capable of partially rescuing the sterile phenotype of a mouse knockout for the homologous gene Dazl.
However, most difficulties in understanding the biological function of DAZ and the genotype-phenotype relation probably arise from the multicopy nature of this gene.
Therefore, only deletions removing the whole of the DAZ gene cluster can be detected, and intragenic deletions or deletions not involving all the DAZ copies, as well as de novo point mutations in affected patients, have yet to be discovered. Therefore, there is still no definitive proof for a requirement of DAZ in spermatogenesis.