Liver zonation
Zonation of parenchymal and nonparenchymal metabolism in liver. 1996
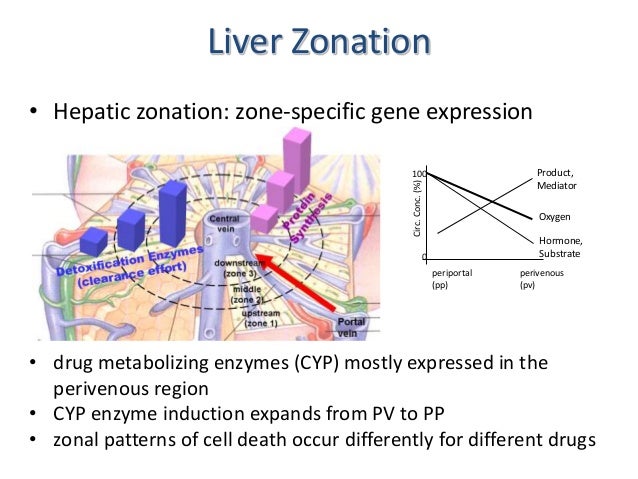
- The enormous number of different liver functions are carried out by parenchymal and four main types of nonparenchymal cells, either alone or in cooperation. Although the liver tissue is uniform on the level of histology, it is heterogenous on the level of morphometry and histochemistry. This heterogeneity is related to the blood supply; cells located in the upstream or periportal zone differ from those in the downstream or perivenous zone in their equipment with key enzymes, translocators, receptors, and subcellular structures and therefore have different functional capacities. This is the basis of the model of metabolic zonation, according to which glucose release from glycogen and via gluconeogenesis, amino acid utilization and ammonia detoxification, protective metabolism, bile formation, and the synthesis of certain plasma proteins such as albumin and fibrinogen occur mainly in the periportal area, whereas glucose utilization, xenobiotic metabolism, and the formation of other plasma proteins such as alpha 1-antitrypsin or alpha-fetoprotein occur predominantly in the perivenous zone. The morphologic and functional heterogeneity is the result of zonal differences in the activation of the cellular genome caused by gradients in oxygen, substrate, hormone, and mediator levels, in innervation, as well as in cell-to-cell and cell-to-biomatrix interactions.


wnt hepatocytes proliferation
Perivenous expression of the mRNA of the three hypoxia-inducible factor a-subunits, HIF1a, HIF2a and HIF3a, in rat liver, 2001
Wnt signalling and the control of cellular metabolism 2010
