 women with AIS (they're 46XY!)
women with AIS (they're 46XY!)
Definition
Androgen insensitivity syndrome (Morris syndrome) is a X-linked genetic condition that risults in the partial or complete inability of the cell to respond to androgens.
the insensitivity to androgens is clinically significant only when it occurs in genetic males. Clinical phenotypes in these individuals range from a normal male habitus with mild spermatogenic defect or reduced secondary terminal hair, to a full female habitus, despite the presence of a Y-chromosome.
It's a rare syndrome that occurs in 1/20.000 born.
AIS represents a spectrum of defects in androgen action and can be subdivided into three broad phenotypes:
- Complete androgen insensitivity syndrome (CAIS), with typical female external genitalia
- Partial androgen insensitivity syndrome (PAIS) with predominantly female, predominantly male, or ambiguous external genitalia
- Mild androgen insensitivity syndrome (MAIS) with typical male external genitalia
Androgen insensitivity syndrome from Wikipedia, the free encyclopedia
Symptoms and diagnosis
The diagnosis of AIS in individuals with a 46,XY karyotype is based on the following clinical findings: undermasculinization of the external genitalia, impaired spermatogenesis with otherwise normal testes, absent or rudimentary müllerian structures, evidence of normal or increased synthesis of testosterone and its normal conversion to dihydrotestosterone, normal or increased luteinizing hormone (LH) production by the pituitary gland, and deficient or defective androgen binding activity of genital skin fibroblasts. Molecular genetic testing of AR, the only gene in which mutations are known to cause androgen insensitivity syndrome, is available clinically and can be used to confirm the diagnosis in some, but not all, instances.
Tests used to diagnose this condition may include blood work to check levels of testosterone , luteinizing hormone (LH), and follicle-stimulating hormone (FSH): they will generally be normal , and we can exclude hormonal disfunction; the genetic test (karyotyping); pelvic ultrasound (search testes in pelvis).
Androgen insensitivity syndrome, 2012. A.D.A.M Medical Encyclopedia
Androgen and androgen receptor: physiological condition
Gonads are histologically distinguishable by 6–8 weeks of gestation. A fetus of that age has both mesonephric (wolffian) and paramesonephric (mullerian) ducts. Subsequent development of one set and degeneration of the other depends on the presence or absence of two testicular hormones: testosterone and AMH. Disruption of typical development may result in the development of both, or neither, duct system, which may produce morphologically intersexual individuals.
Local testosterone causes each wolffian duct to develop into epididymis, vas deferens, and seminal vesicles. Without male testosterone levels, wolffian ducts degenerate and disappear. Müllerian ducts develop into a uterus, fallopian tubes, and upper vagina unless AMH induces degeneration. The presence of a uterus is stronger evidence of absence of testes than the state of the external genitalia.

Androgen is a generic term usually applied to describe a group of sex steroid hormones. Androgens are responsible for male sex differentiation during embryogenesis at the sixth or seventh week of gestation, triggering the development of the testes and penis in male fetuses, and are directed by the testicular determining factor: the gene SRY (sex determining region on Y chromosome) located on the short arm of chromosome Y. The differentiation of male external genitalia (penis, scrotum and penile urethra) occurs between the 9th and 13th weeks of pregnancy and requires adequate concentration of testosterone and the conversion of this to another more potent androgen, dihydrotestosterone, through the action of 5α-reductase in target tissues.
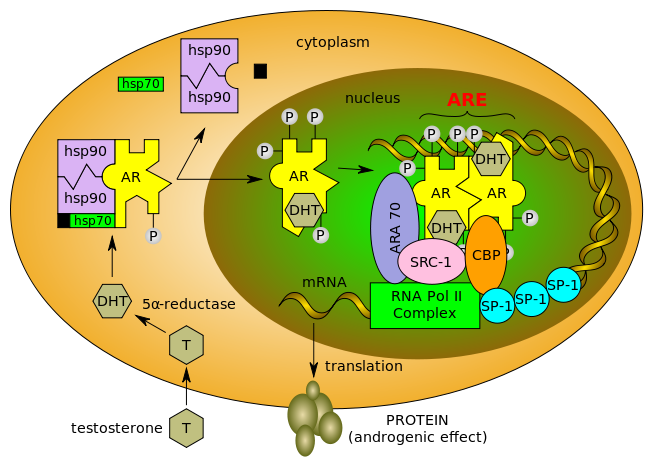
 Testosterone enters the cell and, if 5-alpha-reductase is present, is converted into dihydrotestone (DHT). Upon steroid binding, the androgen receptor (AR) undergoes a conformational change and releases heat-shock proteins (hsps). Phosphorylation (P) occurs before or after steroid binding. The AR translocates to the nucleus where dimerization, DNA binding, and the recruitment of coactivators occur. Target genes are transcribed (mRNA) and translated into proteins.
Testosterone enters the cell and, if 5-alpha-reductase is present, is converted into dihydrotestone (DHT). Upon steroid binding, the androgen receptor (AR) undergoes a conformational change and releases heat-shock proteins (hsps). Phosphorylation (P) occurs before or after steroid binding. The AR translocates to the nucleus where dimerization, DNA binding, and the recruitment of coactivators occur. Target genes are transcribed (mRNA) and translated into proteins.
The AR gene is a protein-coding gene located at Xq11.2-q12. It spans over 90 kb and codes for a protein that functions as a steroid-hormone-activated transcription factor . The androgen receptor (AR), like other members of the nuclear receptor superfamily, has three major functional domains. The AR is characterized by a modular structure consisting of four functional domains: an N-terminal domain (NTD), a DNA-binding domain (DBD), a hinge region, and a ligand-binding domain (LBD; in this case, the ligand being an androgen).

AR Genetic mutations
Individuals XY that present AR genes mutations have a normal production of testosterone and androgens, but cells are not responsive, so the virilization is stopped and the organism (although the individual got Y chromosome) proceed with female differentiation.
The AR NTD is relatively long and displays the greatest sequence variability among nuclear receptors. It is very flexible and displays a high degree of intrinsic disorder. In addition, the NTD has a variable number of homopolymeric repeats, the most important of which is a polyglutamine repeat that ranges from eight to 31 repeats in normal individuals, with an average length of 20 base pairs .
The DBD is centrally located in the AR and it is the most conserved region within the nuclear receptor family. This region has nine cysteine residues, eight of which are involved in forming two zinc fingers, and a C-terminal extension. The first zinc finger, most proximal to the NTD, determines the specificity of DNA recognition, whereas residues in the second zinc finger are involved in AR dimerization. Two AR monomers in a head-to-head conformation bind as a homodimer to androgen response elements which are direct or indirect repeats of the core consensus 5'-TGTTCT-3' or more complex response elements with diverse arrangements of AREs. The C-terminal extension is important for the three-dimensional structure of the DBD and it plays a role in mediating the AR selectivity of DNA interaction.
The hinge region has long been considered to be a flexible linker between the DBD and LBD in the AR. More recently, however, this region was shown to be involved in DNA binding as well as AR dimerization. It was suggested that the hinge region also acts to attenuate transcriptional activity of the AR gene.
As a genetic disorder, AIS presents problems to affected people and their families, and is a major medical challenge for health providers.
Challenges in clinical and laboratory diagnosis of androgen insensitivity syndrome: a case report. 2011
Other causes (not only AR genetic mutations...)
Different factors have been suggested as influencing the expression of AR mutations. The traditional explanation is that the level of competence of co-regulatory proteins acts as a genetic “background” factor in determining the overall clinical outcome. Co-regulators are molecules that interact with nuclear receptors either to increase (co-activators) or to decrease (co-repressors) gene transcription in a ligand-dependent manner by forming a multiprotein complex which involves the basic transcription machinery (Nuclear receptor coactivators: multiple enzymes, multiple complexes, multiple functions. 1999).
Furthermore, some scientific works have recently investigated a patient with PAIS phenotype and defective AR transcription and translation, despite the absence of a molecular abnormality in the entire AR gene. The authors attributed the reduced activity of the AR protein found in this case to the possible existence of a defective AR promoter caused by a mutation or by reduced cellular availability or dysfunction of an AR-promoter-interacting factor critical for the initiation of the AR transcription (Molecular features and clinical phenotypes in androgen insensitivity syndrome in the absence and presence of androgen receptor gene mutations. 2005).
Another probably important mechanism which may account for some variable expressivity is the presence of somatic mosaicism of mutant and wild-type alleles of AR, due to a de novo postzygotic somatic mutation. The proportion of somatic mosaicism in a group of 30 families with AIS patients was relatively high (3 of the 8 patients had de novo mutations) (Inherited and de novo androgen receptor gene mutations: investigation of single-case families. 1998)
Somatic mosaicism should always be considered in AIS individuals with unexpected normal virilization, which could be the result of the expression of the wild-type androgen receptor in some cell lines
It has been previously presented a case of a 46,XY newborn with ambiguous genitalia carrying a mosaic of Val866Met mutation with the wild-type AR gene. Despite the fact that this mutation has usually been associated with the complete AIS phenotype, the newborn exhibited signs of virilization due to the expressed wild-type receptor (Clinical and molecular spectrum of somatic mosaicism in androgen insensitivity syndrome. 1999) .
Discrepancies of phenotype-genotype can also be caused by splice site mutations due to alternative splicing or by differences in 5alpha-reductase activity and thus in adequate DHT availability.
The length of polyglutamine repeats in exon 1 is also a candidate factor leading to diverse phenotypes (Phenotypic variation in a family with partial androgen insensitivity syndrome explained by differences in 5alpha dihydrotestosterone availability. 2001) .
CAIS
The Complete androgen insensitivity syndrome is that condition in which the individual has a complete woman habitus, but is genetically a male, with XY chromosome.
A person with complete AIS appears to be female but has no uterus, and has very little armpit and pubic hair. At puberty, female sex characteristics (such as breasts) develop. However, the person does not menstruate and become fertile.
Androgen insensitivity syndrome, 2012. A.D.A.M Medical Encyclopedia
Management of CAIS women
Areas of management include sex assignment, genitoplasty, gonadectomy in relation to tumor risk, hormone replacement therapy, and genetic and psychological counseling. Individuals with CAIS are raised as females; the assistance of a psychologist experienced in the subject is recommended.
Androgen insensitivity syndrome from Wikipedia, the free encyclopedia
PAIS
Partial androgen insensitivity syndrome is diagnosed when the degree of androgen insensitivity in an individual with a 46,XY karyotype is great enough to partially prevent the masculinization of the genitalia, but is not great enough to completely prevent genital masculinization. This includes any phenotype resulting from androgen insensitivity where the genitalia is partially, but not completely masculinized. Genital ambiguities are frequently
detected during clinical examination at birth, and consequently, a PAIS diagnosis can be made during infancy as part of a differential diagnostic workup.
Pubertal undervirilization is common, including gynecomastia, decreased secondary terminal hair, and / or a high pitched voice. The phallic structure ranges from a penis with varying degrees of diminished size and hypospadias to a slightly enlarged clitoris

Wolffian structures (the epididymides, vasa deferentia, and seminal vesicles) are typically partially or fully developed. The prostate is typically small or impalpable.
During the embryonic stage of development, testes form in an androgen-independent process that occurs due to the influence of the SRY gene on the Y chromosome.
If the testes are located intrascrotally, there may still be significant risk of germ cell malignancy
Impotence may be fairly common; predominantly female phenotypes include a variable degree of labial fusion and clitoromegaly. Ambiguous phenotypic states include a phallic structure that is intermediate between a clitoris and a penis, and a single perineal orifice that connects to both the urethra and the vagina (i.e. urogenital sinus).
MAIS
Mild androgen insensitivity syndrome is the phenotype at the other extreme to CAIS. Genitalia may be underdeveloped for a male or there may be simple coronal hypospadias or a prominent midline raphe of the scrotum. At puberty, MAIS takes two phenotypic forms, both presenting with various degrees of gynecomastia, high-pitched voice, sparse sexual hair and impotence. In one form of MAIS, spermatogenesis and fertility are impaired, while in another spermatogenesis is normal or sufficient to preserve fertility. Although experience with MAIS families is limited, they appear to harbor relatively little phenotypic disparity.
Subjects with MAIS AR mutations may have lower ejaculate volume, higher testosterone levels, higher oestradiol levels and higher androgen sensitivity index. However, the ranges for these variables are highly overlapping between men with and without AR gene mutations.
Androgen insensitivity syndrome: clinical festure and molecular defects. 2008