di Elisa Del Favero e Michela Fringuello
Introduction
Insomnia is a prevalent health complaint, transient insomnia (<2 weeks) affects up to 80% of the population on early basis. Chronic insomnia affects 15% of the population.
This is not simply difficulty with or insufficient sleep. Most insomniac patients feel that the quality of their life, as reflected in their ability to work productively and interact effectively, is seriously compromised by their difficulties with sleep.
Insomnia often ensues from other pathologies , such as cardiovascular diseases (e.g. Angina Pectoris), hormonal imbalance or concurrent psychiatric diseases ; primary insomnia, instead, whose origin is still not known, is uncommon.
Insomnia doesn’t usually need any pharmacological treatment: frequently changing own habits or practising more physical activity may be sufficient.
Traditional treatment for insomnia: benzodiazepines
Despite the availability of effective behavioural treatments for insomnia, pharmacotherapy remains the most widely used treatment method. The most frequently prescribed drugs for insomnia are benzodiazepine-receptor agonists, and, despite the advent of new nonbenzodiazepine agents (zolpidem, zaleplon, zopiclone), traditional benzodiazepines (BZ) are still widely prescribed for sleep.
Benzodiazpines are psycho-active drugs which explicate their action on central nervous system. They are defined as “sedative-hypnotic” drugs: this means that they have two effects:
1. Reduction of anxiety and induction of calmness
2. induction and maintenance of sleep

Hypnotic effect implies a deeper depression of CNS. This aim may be reached by increasing dose of BZD, in fact one of the main characteristics of BZD is that their action is dose-dependant. There may be some difference (in ratio “drug dose/importance of depression”) among different types of these drugs. For example, drug A (Fig.1) is a more powerful reducing agent than B, which needs bigger amount to reach the same level of nervous depression. This last is the case of BZD.
Chemical classification
The core chemical structure of BZD derives from the fusion of a benzene ring and a diazepine ring. In order to have a sedative activity, a halogen or a nitrogroup are required in position 7.
Fig.2: chlordiazepoxide

Pharmacokinetics
BZD are administered per os; the absorption rate depends especially on drug's liposolubility. This feature is fundamental to cross the blood-brain barrier, but makes the drug able to cross the placenta in pregnant women, and this is obviously dangerous for fetal CNS development.
Clearance of BZD occurs in liver, where the drug metabolized by cytochrome P450 (CYP450) enzymes (stage I). After that, metabolites are conjugated in order to form glucuronides (stage II), which are excreted in urine. Despite this, there are metabolites from stages I which are pharmacologically active and have a longer half-life than the primary compounds. Many drugs, including oral contraceptives, some antibiotics, antidepressants, and antifungal agents, inhibit cytochrome enzymes in the liver. They reduce the rate of elimination of the benzodiazepines that are metabolized by CYP450, leading to possibly excessive drug accumulation and increased side-effects. In contrast, drugs that induce cytochrome P450 enzymes, such as the antibiotic rifampicin, and the anticonvulsants carbamazepine and phenytoin, accelerate elimination of many benzodiazepines and decrease their action.
A benzodiazepine can be placed into one of three groups by its elimination half-life, or time it takes for the body to eliminate half of the dose.
• Short-acting compounds have a median half-life of 1–12 hours. They have few residual effects if taken before bedtime, rebound insomnia (=the rebound effect, is the emergence or re-emergence of symptoms that were either absent or controlled while taking a medication, but appear when that same medication is discontinued, or reduced in dosage. In the case of re-emergence, the severity of the symptoms are often worse than pretreatment level) may occur upon discontinuation, and they might cause daytime withdrawal symptoms such as next day rebound anxiety with prolonged usage. Examples are brotizolam and triazolam.
• Intermediate-acting compounds have a median half-life of 12–40 hours. They may have some residual effects in the first half of the day if used as a hypnotic. Rebound insomnia, however, is more common upon discontinuation of intermediate-acting benzodiazepines than longer-acting benzodiazepines. Examples are alprazolam, lorazepam and temazepam
• Long-acting compounds have a half-life of 40–250 hours. They have a risk of accumulation in the elderly and in individuals with severely impaired liver function, but they have a reduced severity of rebound effects and withdrawal. Examples are diazepam, clorazepate and flurazepam.
Benzodiazepine effects on sleep disease
The two types of benzodiazepine, short and long- acting , shows different effects on total sleep time and number of awakenings . Both of these compounds significantly
1. decreased the number of awakenings and
2. increased total sleep time with one week of nightly administration.
While the short-acting compounds showed significant hypnotic effects already after a single night administration, long-acting compounds need multiple nights of drug administration, and therefore drug accumulation, to achieve significant hypnotic efficacy.
Even if use of benzodiazepine helps to increase total sleep times, the architecture of sleep (composed by 4 NonREM stages, of which the 1 and2 ones are considered light-sleep phases, while 3 and4 phases are associated with deep-sleep, and REM stage, that is thought to be the most restful phase of sleep) is significantly altered.
One of the most surprisingly result about use of both short- and long-acting benzodiazepine is that they increased the latency to REM sleep. In addiction benzodiazepine administration is associated with a significant decrease in the percentage of REM, especially the long-acting ones. These findings demonstrate that although both compounds affected REM latency, drugs with the longest half-life have a more powerful capacity to suppress the total percentage of REM across the night.
It is also noticed that although neither compound significantly affected the latency to stage 3-4, long-acting drugs significantly suppressed the total percentage of stage 3-4 (sleep phases characterized by delta slow waves), benzodiazepine users spend less time in Stage 3 and 4 sleep, and more time in stage 2, characterized by only theta waves , not indicative of deep sleep.
The passage from theta (phase2) to delta frequencies (phases 3, 4) is due to progressive hyperpolarisation of the thalamocortical cell membranes. These suggestions might imply that chronic BZ use is very disturbing to the EEG and, especially, by affecting the hyperpolarization process.
Pharmacodynamics
BZD binds the GABAa receptors. This receptor is ligand-gated ion (Chlorine, in this case) channel, also known as ionotropic receptor that, when activated, induces cellular hyperpolarization. There are also GABAb receptors, that are instead G protein-coupled receptors or metabotropic receptors. These ones, anyway, are not bound by BZD. This is due to a lower affinity.
GABAa receptor, which is located on post-synaptic membrane,is a pentameric transmembrane receptor that consists of five subunits arranged around a central pore. There are numerous subunit isoforms for the GABAA receptor, which determine the receptor's agonist affinity, chance of opening, conductance, and other properties. In humans, the units are:
• 6 types of α subunits ( α1, α2, α3, α4, α5, α6)
• 4 types of β subunits
• 3 types of γ subunits
• and other subunits named δ, ε, and π (less important for this discussion).
One of the most common isoform of GABAa receptor in CNS is formed by:
• 2 α1 subunit
• 2 β2 subunit
• 1 γ2 subunit
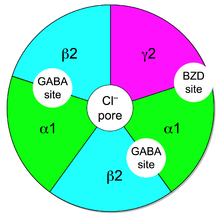
In this isoform there are two sites for GABA between a α1 and β2 subunits; the site for BZD is between a α1 subunit and the γ2 subunit.
BZD enhance GABAergic neurotransmission on the whole CNS. They have an allosteric positive effect on post-synaptic inhibition induced by GABA. Taking benzodiazepines with alcohol, opioids and other central nervous system depressants potentiates their action, beacuse these substances bind GABA receptors in different sites than BZP. This often results in increased sedation, impaired motor coordination, suppressed breathing, and other adverse effects that have potential to be lethal.
What is BZD effect on organs?
There are basically 6 effects mediated by BZD:
1. SEDATION
2. HYPNOSIS
3. ANAESTHESIA
4. ANTICONVULSANT EFFECT
5. MIORELAXANT EFFECT
6. DEPRESSION OF BREATHING AND CARDIOVASCULAR FUNCTIONS
7. bearable ANTEROGRADE AMNESIA
In this dissertation, we are especially intrested in the hypnotic effect.
By definition, all sedative-hypnotic drugs induce sleep when administered in adequately high dose. General effects of BZD on sleep stages are:
• decrease in sleep latency
• increase in stage 2 duration
• reduction in stage 4 and REM sleep duration
Why Benzodiazepines induced sleep is not the same as natural sleep
The faster onset of sleep represents the most useful clincal score; despite this, experiments demonstrate that BZD-induced sleep is not as restful as natural sleep. This is probably due to reduction of slow-wave sleep (stage 4) and decrease in REM sleep.
It is of fundamental importance to notice that Benzodiazepines are REM sleep-suppressant medications for two reasons:
1. REM sleep is known to be the “restful” phase of sleep and
2. it is known to have a role in learning and memory consolidation.
Consequences on memory and learning
The acquisition and consolidation of new information (learning) is related to quantity of REM sleep because sleep spindles (particular desynchronized frequency in sleep waves)seem to reflect the activity of thalamocortical neurons, that are involved in development of long term potentiation of synapses, linked to learning and memory. A bigger quantity of sleep spindles is noticed in the night subsequent to an intensive motor or cognitive training. Similar effects result from study of PGO (ponto-geniculo-occipital) waves that are related to rapid eye movement of REM sleep. It is shown that PGO traces are increased during night (in particular in REM stage) when anyone had received cognitive stimulation (for example anything to learn)during the wake. It could be related with a cortical activity of storing new information

REM sleep burst neurons, PGO waves, and eye movement information; J. P. Nelson, R. W. McCarley, and J. A. Hobson, 1983
As were shown in the research EFFECTS OF BENZODIAZEPINES ON SLEEP AND WAKEFULNESS; T. Roth, F. Zorick, Jeanne Sicklesteel, and E. Stepanski, 1981
use of benzodiazepine affect many daily activities and types of tasks, in particular cognitiveness, vigilance, and memory usually in the form of performance decrement.
It is maybe due to the interference with hyperpolarisation caused by benzodiazepine assumption. In facts, as we have said, Benzodiazepine acts binding allosteric site on GABA A receptor, increasing its affinity for GABA and potentiating its effect.
This causes a decrement of excitability of thalamocortical neuronal cells, that aren’t’ able to generate action potential.
The nature of the impairment is related to two pharmacological properties of the drug. First, the duration of the decrement is related to the half-life of the drug. Second, the degree of performance decrement is related to the hypnotic potency of the drug.
Consequences on sleep quality
The second consequence due to the decrement of REM sleep quantity is the worsening of sleep quality. REM has the peculiar property to be a restful sleep primarily because in this phase of the sleep all voluntary muscles (except eye muscles as its name suggests) have no tonic activity and so are totally paralyzed (it is thought to be a protective behaviour against dangerous movements during a non conscious phase of our life).
The paralysis is caused by an inhibitory signal, mediated by neurotransmitter GABA, that turns off some specific neuronal cells.
Identification of the Transmitter and Receptor Mechanisms Responsible for REM Sleep Paralysis; J.Neurosci, 2012
Previous studies shown that ionotropic GABA receptors (GABA A receptor)are involved in this mechanism, even if the blockage of this type of receptor is not enough to avoid the paralysis: both ionotropic (GABA A) and metabotropic (GABA B) receptor have to be blocked not to have the paralysis appeared and to notice high levels of muscular activity. These results demonstrate that multiple receptor mechanisms trigger REM sleep paralysis.
It could means that to maintain a superior motor control (in this case no voluntary muscular activity), the two neurotransmitters have to act togheter. Neither metabotropic nor ionotropic receptor mechanisms alone are sufficient for generating REM paralysis.
Benzodiazepine indeed act only on GABA A receptor while they have no effect on GABA B metabotropic receptor because these last ones have no allosteric sites for benzodiazepine. It could be the reason of incapacity of benzodiazepine to bring to a complete REM sleep. In this case muscular activity is not completely cut off, not allowing muscular paralysis, and so not guaranteeing a restful sleep.