DEFINITION
Albinism, derived from the Latin, albus, meaning white, is a group of inherited disorders in which melanin biosynthesis is reduced or absent and cause complete or partial absence of pigment in the skin, hair or eyes. Albinism: Classification, Clinical Characteristics and Recent Findings, 2009
Other definitions, according to the Diseases Database, are:
• General term for a number of inherited defects of amino acid metabolism in which there is a deficiency or absence of pigment in the eyes, skin, or hair.
• A group of genetic conditions marked by little or none of the pigment melanin in the skin, hair, and/or eyes. People with albinism may have vision problems and white or yellow hair; reddish, violet, blue or brown eyes; and pale skin.

 Oculocutaneous Albinism
Oculocutaneous Albinism
CLASSIFICATION OF ALBINISM
Several genes have been found to be responsible for albinism. The current classification of albinism is determined by the affected gene, making the previously used terms, “partial or complete” and “tyrosinase-positive or tyrosinase-negative” obsolete.
• The Oculocutaneous albinism ( Oculocutaneous Albinism) is a group of four autosomal recessive disorders caused by either a complete lack or a reduction of melanin biosynthesis in the melanocytes. The different types of OCA are caused by mutations in different genes but the clinical phenotype is not always distinguishable, making molecular diagnosis a useful tool and essential for genetic counseling. Albinism: Classification, Clinical Characteristics and Recent Findings, 2009
The gene for tyrosinase ( TYR ) on chromosome 11q14-21 and the P gene on chromosome 15q11.2 are the most commonly affected genes; mutations on these genes cause oculocutaneous albinism type 1 (OCA1) and oculocutaneous albinism type 2 (OCA2), respectively.
Oculocutaneous albinism, 2007
• Another type of albinism, caused by mutations on Xp22.3, is the ocular albinism (OA1 ), which affects males because of X-linked inheritance;
• Other types of albinism occur more infrequently, including those associated with systemic manifestations, such as Hermansky-Pudlak Syndrome ( Hermansky-Pudlak Syndrome; bleeding disorder due to absence of dense bodies in platelets) and Chédiak Higashi syndrome (immunodeficiency and neurologic problems).
Albinism: Classification, Clinical Characteristics and Recent Findings, 2009

The TLDA or Tyrosinase Lack-Depending Albinism, in which there is the mutations of the tyrosinase gene ( TYR ), results in OCA1.
OCA1 is further divided into OCA1A and OCA1B according to the residual activity of the Tyrosinase enzyme:
• OCA1A is the classic form of tyrosinase-negative OCA, resulting in a life-long absence of melanin pigment formation in the affected individual;
• OCA1B : affected individuals may accumulate varying amounts of pigment throughout their life, including the ability to tan, depending on the severity of the mutation and their ethnic background. In some cases, OCA1B has been incorrectly diagnosed as autosomal recessive OA or not diagnosed as albinism at all due to the patient’s near-normal visual acuity. In individuals with OCA1B, the ocular defect are not as severe as in individuals with OCA1A. Oculocutaneous albinism, 2007
EPIDEMIOLOGY
Albinism can affect people of all ethnic backgrounds and has been extensively studied. Approximately one in 17,000 people have one of the types of albinism. This suggests that about 1 in 70 people carry a gene for OCA. Prevalence of the different forms of albinism varies considerably worldwide, partly explained by the different founder mutations in different genes and the fact that it can be difficult clinically to distinguish between the different subtypes of albinism among the large normal spectrum of pigmentation. OCA2 is the most prevalent form worldwide.
In particular, OCA1 is transmitted in an autosomal recessive manner and it is expressed in males and females with the same frequency.
OCA1 has a prevalence of approximately 1 per 40,000 in most populations, but is very uncommon among African-Americans. Oculocutaneous albinism, 2007
SYMPTOMS
The eye and optic system abnormalities are common to all types of albinism (OCA and OA) and are probably related to the reduction of melanin during embryonic development and early postnatal life. Characteristic changes in the optic system include:
• reduced pigmentation of the iris (iris translucency) and of the retinal pigment epithelium
• foveal hypoplasia
• decreased visual acuity usually in the range 20/60 to 20/400
• misrouting of the optic fibers at the chiasm
• nystagmus, strabismus
• refractive errors
• degree of color vision impairment (sometimes)
Molecular and Clinical Characterization of Albinism in a Large Cohort of Italian Patients, 2011
Photobia may characterize the disease of some patients, sometimes even in a very important way.
Iris translucidity is demonstrable by slit lamp examination.
One of the features that is mainly linked to OCA is the misrouting of the optic fibers, consisting in an excessive crossing of the fibres in the optic chiasma, which can result in strabismus and reduced stereoscopic vision. The abnormal crossing of fibres can be demonstrated by monocular visual evoked potential. Absence of misrouting excludes the diagnosis of albinism.
Delayed visual maturation has been reported in albinism. It is not unusual for parents of an infant with albinism to note poor fixation on faces and objects and a delay in visual development.
Nystagmus (involuntary, rapid, rhythmic movement of the eyeball) typically develops by 6 to 8 weeks of age and is initially slow with a large amplitude, but the amplitude typically decreases within the first year of life.
The degree of skin and hair hypopigmentation, when present, varies along a wide clinical spectrum of severe to mild phenotypes. The clinical spectrum of OCA varies both within and among genotypes. However in general:
• In OCA1A the hair, eyelashes and eyebrows are white, and the skin is white and does not tan. Irises are light blue to almost pink, and fully translucent. Pigment does not develop and amelanotic nevi may be present. The symptoms do not vary with age or race. Visual acuity is 1/10 or less, and photophobia is intense.
• In OCA1B, the hair and skin may develop some pigment with time (after 1 to 3 years), and blue irises may change to green/brown. Temperature-sensitive variants manifest as having depigmented body hairs, and pigmented hairs on hands and feet due to lower temperatures. Visual acuity is 2/10. This phenotype was previously known as yellow albinism.
• The other OCAs have a mild symptomatology, although OCA2 may reach a visual acuity of 3/10. The skin pigmentation is reduced but not as reduced as OCA1A, and the degree of pigmentation is very variable.
OCA2 is characterized by light brown hair and skin, and gray irises in Africans, while OCA3 is associated with the red pigmentation of the hair and the reddish-brown skin (xanthism). The OCA3 doesn't show visual anomalies because the hypopigmentation is so mild that the correct development of the optical system is saved.

Caused by the reduction or the lack of the production of skin pigment, patients with albinism (overall with OCA1A) have an increase of the incidence of skin cancer. Oculocutaneous albinism, 2007
DIAGNOSIS
The diagnosis of OCA is based on clinical findings of hypopigmentation of the skin and hair, in addition to the characteristic ocular symptoms.
A study showed that patients with albinism had a corneal reflex deviated nasal to the center of the pupil, known as a positive angle kappa. Prism and alternate cover test at a more rapid rate than usual (to diminish effect of increased nystagmus amplitude with monocular occlusion) often discloses strabismus. Because of the positive angle kappa, an esotropia may be masked or appear diminished in amount on causal gaze, compared with the measured deviation, whereas an exotropia may appear larger than measured.
Albinism: Classification, Clinical Characteristics and Recent Findings, 2009
To value the Iris Transillumination is used a tonometer, a machine that directs a small bright light through the pupil of the patient and let the examiner to fixate on the iris. Iris transillumination is best performed in a dark room after the examiner has become adapted to the darkness. The amount of iris transillumination can vary and a grading scheme has been described:
• grade 1, punctate areas of transillumination, indicating that a marked amount of pigment is present in the posterior iris epithelium;
• grade 2, moderate iris pigment;
• grade 3, minimal iris pigment;
• grade 4, full transillumination of the iris because of the absence of melanin pigment
Albinism: Classification, Clinical Characteristics and Recent Findings, 2009
Examination of the fundi typically shows that foveal development is absent. A few patients with albinism, who have vision 20/50 or better, have some rudimentary foveal development, and some thinning of the retina in the foveal area has been demonstrated with optical coherence tomography.

a) fundus picture of a patient with albinism
b) fundus picture of a normal eye
The appearance of the macula has been graded as follows:
• grade 1, choroidal vessels easily seen in macula;
• grade 2, choroidal vessels less distinctly seen because of translucent retinal pigment epithelium;
• grade 3, opaque macula so that choroidal vessels are not visible.
These grading scales for the iris and macula can be useful in clinical studies to more precisely describe the study population because these characteristically persist over time. In addition, careful inspection can show granular melanin pigment in the macula in a few patients with albinism and occasionally finely granular pigment has been identified beyond the macula. The
presence of melanin pigment in the macula correlates with better visual acuity.
Albinism: Classification, Clinical Characteristics and Recent Findings, 2009
Due to the clinical overlap between the OCA subtypes, molecular diagnosis is necessary in order to establish the gene defect and thus the OCA subtype. Molecular genetic testing of TYR and OCA2 are available on a clinical basis, while at present, analysis of TYRP1 and MATP is on research basis only. Molecular genetic testing is based on mutational analysis of the genes, by standard screening methods such as denaturing high performance liquid chromatography (DHPLC) or single stranded conformational polymorphism (SSCP), followed by DNA sequencing. Oculocutaneous albinism, 2007
Genetic counseling and antenatal diagnosis
Carrier detection and prenatal diagnosis are possible when the disease causing mutations have been identified in the family. Both disease causing mutations in an affected person have to be identified and established to be on the paternal and maternal chromosome, respectively, before prenatal diagnosis can be performed in pregnancies at 25% risk for an affected child. The testing can be done on DNA extracted from chorion villus sampling (CVS) at 10–12 weeks gestation or on DNA extracted from cultured amniocytes. Preimplantation diagnosis using molecular genetic analysis is also possible in principle, but to our knowledge, this has not been carried out.
Requests for prenatal diagnosis for OCA are not common, and may reflect the nature of the condition (not affecting intellectual functions or general health). Oculocutaneous albinism, 2007
PATHOGENESIS
Introduction
Albinism is a disease in which there is a reduction of the production of pigment in the skin, hair and eyes. This pigment is called Melanin which is a large biopolymer derived from the progressive oxidation of the amino acid Tyrosine in the absence or in the presence of sulfhydryl groups from cysteine, giving rise to black-brown (*eumelanin*) or yellow-red (*pheomelanin*) compounds, respectively. Its biosynthesis occurs in specialized cells called Melanocytes and retinal pigment epithelium (RPE). The former derive embryologically from the neural crest and then migrate in several organs and tissues, including the basal layer of the epidermis, where they play a critical role in photo-protection and camouflage, the eye (choroid and iris stroma), the inner hear, and the leptomeninges. In contrast, the RPE derives from the neuroectoderm, similarly to the neurosensory retina, and is situated behind the photoreceptor layer in the posterior segment of the eye, but also extends anteriorly to form the innermost part of the iris, becoming the iris pigment epithelium (IPE). The RPE is implicated in photo-absorption, provides structural and functional support to photoreceptors, and - last but not least - plays a fundamental role in the development of the retina and visual pathways.
Signaling pathways in melanosome biogenesis and pathology, 2010

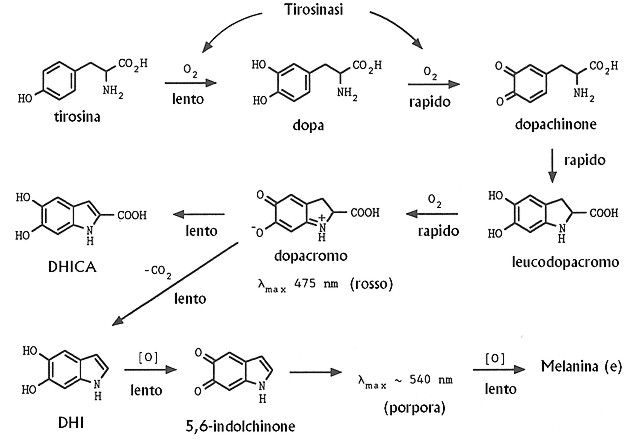
In the OCA the synthesis of melanin is reduced or even absent, because of the alteration of the activity of the Tyrosinase enzyme. This alteration is caused by mutations into the TYR gene (OMIM 606933)
Tyrosinase is localized on chromosome 11q14-21 and the coding region is divided into five exons spanning more than 65 kb that codes for a polypeptide of 529 aminoacids.
Tyrosinase is a copper-containing enzyme that catalyzes at least the first two reactions in melanin biosynthesis: the hydroxylation of tyrosine to L-3,4-dihydroxyphenylalanine (DOPA) and the subsequent oxidation of the nascent DOPA to DOPAquinone.
Tyrosinase has also been reported to catalyze a third, distal step in the melanin biosynthetic pathway, the oxidation of 5,6-dihydroxyindole (DHI) to indole-5,6-quinone, although its role in this latter reaction has never been directly confirmed. So Tyrosinase has 3 different activities: Tyrosine Hydroxylase, DOPA oxidase and DHI oxidase. Mutational Mapping of the Catalytic Activities of Human Tyrosinase, 1992
The two steps occurr during stages III and IV of melanosome maturation. During stages I and II of melanosome biogenesis, intralumenal fibril is deposited in immature melanosome vesicles.11 Melanin synthesis begins when tyrosinase is delivered through the early endosome to the melanosome sorting pathway to stage III melanosomes. Melanin synthesis terminates with the complete blackening of stage IV melanosomes. Signaling pathways in melanosome biogenesis and pathology, 2010

Schematic representation of TYR sorting through ER and Golgi.
Panel Ai shows the ER processing of TYR— (1) the nascent peptide is translocated within the ER lumen through the sec61 translocon and BiP binding occurs transiently, the protein becomes glycosylated with the addition of the glycan moiety. That is trimmed later to facilitate the lectin chaperone binding;
(2) calnexin (CNX) binding occurs when two trimmed glycans (Glc1Man9GlcNAc2) are added to the polypeptide chain and (3) calreticulin (CRT) binding is initiated with the addition of the 3rd glycan.
ERp57 being associated with CNX or CRT supports the disulfide bond formation;
(4A) properly folded TYR is homodimerized, although hetero-oligomeric interactions cannot be ruled out and (5) then the protein exits ER to reach Golgi.
However, the un/misfolded protein re-enters the CNX/CRT cycle via UGGT (UDP-glucose: glycoprotein glucosyl transferase) (4B) and if still fails to fold correctly, is sorted out to the cytosol by EDEM (ER degradation enhancing—mannosidase-like protein) for proteasomal degradation (4C).
Panel Aii demonstrates the role of glycan-trimming during the ER maturation of TYR. Different forms of glycans shown are as follows: Glc3 form, Glc3Man9GlcNAc2; Glc2 form, Glc2Man9GlcNAc2 and Glc1 form, Glc1Man9GlcNAc2.
Panel B schematically demonstrates the Golgi processing of TYR— (1) transport of ER-processed TYR (correctly folded) via CopII vesicles to the cis-Golgi; (2) further modification of TYR within Golgi, followed by (3) Cu-loading, which may occur directly through Menkes copper transporter (MNK) or may be aided by a carrier protein; (4) sorting out of matured TYR from trans Golgi network (TGN) to stage II melanosome via direct or indirect pathway. Tyrosinase and ocular diseases: Some novel thoughts on the molecular basis of oculocutaneous albinism type 1, 2007
When the mutations of the TYR gene changes the correct sequence of the amminoacids of Tyrosinase, a mis/unfolded protein is producted and this has not enzymatic activity. So it is linked to EDEM protein and directed to the proteasoma where is degradeted. For this reason melanosomes can't mature and melanin is not produced.
Mutations completely abolishing tyrosinase activity result in OCA1A, while mutations rendering some enzyme activity result in OCA1B allowing some accumulation of melanin pigment over time. Almost 200 mutations in TYR are known. As with all recessive disorders, the "mildest" mutation is determining for the phenotype.
Mutational Mapping of the Catalytic Activities of Human Tyrosinase, 1992
Oculocutaneous albinism, 2007
Mutations, in OCA1, can affect, in different way, the three catalytic activities of Tyrosinase. This indicates that, not surprisingly, sequences that can influence these activities are distributed throughout the length of the tyrosinase polypeptide. However, although the various catalytic activities of tyrosinase may well share at least some structural elements of the tyrosinase polypeptide, data strongly suggest that they also involve distinct sequences. The DOPA oxidase and DHI oxidase activities of normal tyrosinase are temperature-sensitive, and essentially all of the mutations tested resulted in virtually identical effect on both these activities. These two reactions are chemically rather similar, and it seems likely that they involve a single catalytic site. In contrast, the tyrosine hydroxylase activity of the normal tyrosinase is thermostable, and at least one of the mutations tested, Ala-Thr, exerted divergent effects on the tyrosine hydroxylase versus the DOPA/DHI oxidase activities. This suggest that the catalytic site for tyrosine hydroxylase activity of the tyrosinase may be distinct from that for the DOPA/DHI oxidase activity. Mutational Mapping of the Catalytic Activities of Human Tyrosinase, 1992
Melanin Function and Pathology
Melanin is involved in some activities:
1. Protection of the skin: melanosomes, containing melanin, distributes around the cell nuclei of the skin to protect them to UV Radiations. UVR infact can produce Reactive Oxigen Species that are involved in oncogenesis and produce the break of the folate into cells by photolysis. The folate is very important for the cell division.
Human skin pigmentation as an adaptation to UV radiation, 2010
Signaling pathways in melanosome biogenesis and pathology, 2010
Melanin, overall Eumelanin, produces tanning, which is a mild sunscreen, protecting the skin from photodamage and cancer.
2. Protection of the eye: besides the role of pigmentation during ocular development, melanin also acts as a filter, by modulating the proper scattering of incoming photons to the eye, and protecting the retina from light-damage.
3. Correct Development: Melanin lets the correct development of the eye in particular of the Fovea and direct fate of ganglion retinal cell axons pathfinding, preventing the misrouting of the optic fibers.
Further, its deficiency is associated with uncontrolled pendular eye movements (nistagmus)
Signaling pathways in melanosome biogenesis and pathology, 2010
AAV-mediated Tyrosinase Gene Transfer Restores Melanogenesis and Retinal Function in a Model of Oculo-cutaneous Albinism Type I (OCA1), 2011
THERAPY
Management of eye problems
Reduced visual acuity can be helped in various ways. Clinics specialized in low vision will provide the expertise. Glasses, possibly bifocals, may often be of sufficient help. Photophobia can be helped with dark glasses or photocromic lenses that darken with exposure to bright light. Nystagmus may be helped with contact lenses. For strabismus it may be necessary to patch one eye in children to force the non-preferred eye to be used.
Children should be given special attention at school, for instance with high contrast written material, large type textbooks, various optic devices as enlargement machines (closed circuit TV), and the use of computers. Oculocutaneous albinism, 2007
Surgery can help patients with residual Nystagmus. In fact the amplitude of nystagmus diminishes as the child matures and in some cases may be detected clinically only as latent nystagmus with monocular occlusion in older children and adults.
The main problem is that patients often develop an anomalous head posture to damp the nystagmus, referred to as the null point, where visual acuity is usually improved. When the head posture is sufficiently large and stable in amount, surgery can transfer the null point to a position closer to primary gaze with a extraocular muscle surgery (Kestenbaum-Anderson procedure).
Another surgical procedure (retroequatorial recession of the horizontal rectus muscles) has been described to improve visual acuity in patients with nystagmus due to many etiologies.
Most recently, tenotomy of the horizontal rectus muscles with reattachment at the original insertion has been introduced to improve visual acuity and calculated acuity function.
The results are intersting and over the resolution of the Nistagmus, there has also been reported improvements in the visual acuity. Thanks to this some patient were allowed to obtain a driver’s license so surgery should at least be discussed with the patient, in addition to other potential methods to improve visual function.
Albinism: Classification, Clinical Characteristics and Recent Findings, 2009
Skin
Most people with severe forms of OCA do not tan and easily get sunburned. Those forms with a little pigment developing with age may not be very bothered by the sun. Sunscreens are recommended with at least a sun protection factor of 15. Ultraviolet rays can penetrate light T-shirts especially when wet. Now, T-shirts have been developed which protect against the sun even when wet. The incidence of skin cancer is increased in patients with OCA. Oculocutaneous albinism, 2007
Future Treatments
Some studies have reported interesting results which could open new frontiers in the treatment of the albinism.
Below, two studies are citated:
1. Nitisinone improves eye and skin pigmentation defects in a mouse model of oculocutaneous albinism: Ighovie F. Onojafe, David R. Adams, Dimitre R. Simeonov, Jun Zhang, Chi-Chao Chan, Isa M. Bernardini, Yuri V. Sergeev, Monika B. Dolinska, Ramakrishna P. Alur, Murray H. Brilliant, William A. Gahl and Brian P. Brooks, 2011.
This study examines the reaction of OCA1 mice to Nitisinone, an inhibitor of an enzyme of the catabolic pathway of the tyrosine. Administering Nitisinone in OCA1 mice, blood levels of Tyrosine increased, followed by a reprise of the pigmentation in OCA1B mice, there was a residual activity of Tyrosinase in. There was also an improvement of the visual acuity, directly collected to the precocity of the treatment. Earlier the treatment, greater the inprovement.
2. AAV-mediated Tyrosinase Gene Transfer Restores Melanogenesis and Retinal Function in a Model of Oculo-cutaneous Albinism Type I (OCA1): Annagiusi Gargiulo, Ciro Bonetti, Sandro Montefusco, Simona Neglia, Umberto Di Vicino, Elena Marrocco, Michele Della Corte, Luciano Domenici, Alberto Auricchio, and Enrico M Surace.
This study has reported a reprise of the melanization and the production of eye pigment in mice cured with subretinal injections of TYR gene by a vector ( AAV2/1-CMV-_hTYR_ is a ubiquitous Citomegalovirus harboring the TYR gene).
Bibliografy
- Albinism: Classification, Clinical Characteristics and Recent Findings; C. Gail Summers, 2009
- Molecular and Clinical Characterization of Albinism in a Large Cohort of Italian Patients; Annagiusi Gargiulo, Francesco Testa, Settimio Rossi, Valentina Di Iorio, Simona Fecarotta, Teresa de Berardinis, Antonello Iovine, Adriano Magli, Sabrina Signorini, Elisa Fazzi, Maria Silvana Galantuomo, Maurizio Fossarello, Sandro Montefusco, Alfredo Ciccodicola, Alberto Neri, Claudio Macaluso, Francesca Simonelli and Enrico Maria Surace, 2011
- Mutational Mapping of the Catalytic Activities of Human Tyrosinase; Ram K. Tripath, Vincent J. Hearing, Kazunori Urabe, Pilar Aroca and Richard A. Spritz, 1992 [5]
- Oculocutaneous albinism; Karen Grønskov, Jakob Ek and Karen Brondum-Nielsen, 2007
- Tyrosinase and ocular diseases: Some novel thoughts on the molecular basis of oculocutaneous albinism type 1; Kunal Ray, Moumita Chaki, Mainak Sengupta, 2007
- Human skin pigmentation as an adaptation to UV radiation; Nina G. Jablonski and George Chaplin, 2010
- Signaling pathways in melanosome biogenesis and pathology; Maria Schiaffino, 2010
- AAV-mediated Tyrosinase Gene Transfer Restores Melanogenesis and Retinal Function in a Model of Oculo-cutaneous Albinism Type I (OCA1): Annagiusi Gargiulo, Ciro Bonetti, Sandro Montefusco, Simona Neglia, Umberto Di Vicino, Elena Marrocco, Michele Della Corte, Luciano Domenici, Alberto Auricchio, and Enrico M Surace
- Nitisinone improves eye and skin pigmentation defects in a mouse model of oculocutaneous albinism: Ighovie F. Onojafe, David R. Adams, Dimitre R. Simeonov, Jun Zhang, Chi-Chao Chan, Isa M. Bernardini, Yuri V. Sergeev, Monika B. Dolinska, Ramakrishna P. Alur, Murray H. Brilliant, William A. Gahl and Brian P. Brooks, 2011