Federica Castagneris
DEFINITION
Parkinson's disease is the second most common neurodegenerative disease, after Alzheimer; affecting approximately 1% of inividuals olther then 60 years. It's a type of motor system disorder that results from the progressive loss of dopaminergic neurons in Substantia Nigra pars compacta (SNpc) of the midbrain and, consequently, striatal dopaminergic deficiency.
So Parkinson disease is predominantly a disorder of the basal ganglia which are a group of nuclei situated at the base of the forebrain. The striatum, composed of the putamen and the caudatum, is the largest nuclear complex on the basal ganglia; it receives imput from several areas of the cerebral cortex as well as inhibition and excitatory ones from the dopaminergic cells of the SNpc depending on the type of the hydroxitriptamine receptors expressed by two different class of striatal neurons: those that project directly to the internal segment of the Globus Pallidus (GPi),the major output site of the basal ganglia that belong to the DIRECT PATHWAY and those that project to the external segment of the GP, enstablishing an INDIRECT PATHWAY to the GPi via the subthalamic nucleus.
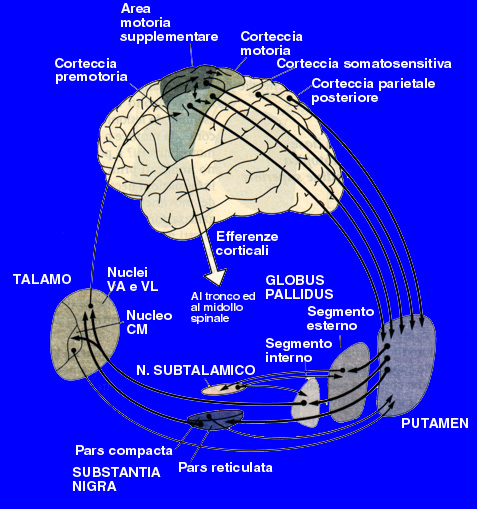
Tha action of the direct and indirect pathways regulate the neuronal output form the GPi, which provides tonic inhibitory imput to the thalamic nuclei projecting to the primary and supplementary motor areas. Signals from the cortex are processed through the basal ganglia thalamocortical motor circuit and retourn to the same areas via a feedback pathway modulating tha cortical output necessary for normal movement.
For this reason the increased inhibition of the thalamocortical pathway suppresses voluntary actions while other spontaneous movement tends to appear,no more down-regulated.
SYMPTOMS & DIAGNOSIS
The most common clinical findings of Parkinsonism are tremor,rigidity and bradikinesia, all used for enstablishing the diagnosis, but it can only be confirmed through the documentation of the death of dopaminergic cells in SNpc (up to 70%) with intracytoplasmatic inclusions, the so-called "Lewy bodies", in the remaining intact nigral neurons.
Lewy bodies are the pathological hallmark of Parkinson disease and the disribution of them is often directly related to the expression and digree of the clinical symptoms that can be monitorized by test such as neuroimaging.
Even if the cause is nowadays not so clear, it seems that mutations in specific genes play an important role; these genes codes for fibrillar alpha-synuclein, a protein that is the main component of Lewy bodies. A new hypothesis is that PD-linked proteins' aggragates could be implicated in the activation of microglial cells to induce a chronic and sustained neuroinflammation involved in PD pathogenesis. Within the microenvironment of the brain, glial cells play a critical role in homeostatic mechanism promoting neuronal survival: microglia have a specialized immune-surveillance role and mediate innate responses to invaliding pathogens by secreting a myriad of factors that include cytokines, chemokines, reactive oxigen, nitrogen species and growth factors. Some of these have neuroprotective and trophic activities and in brain aid repairment processes, while others trigger apoptotic cascades in neurons.
Therefore pro- and anti-inflammatory responses must be in balance to prevent the potential detrimental effects of prolonged and unregulated inflammation-induced stress on vulnerable nigral neuronal population leading to death and predisposing an individual to development of PD.


So evidences suggested that alpha-synuclein's inclusions elicite the activation of resident microglia in specific subcortical regions: the inflammation-derived oxidative stress and cytokine-dependent toxicity may contribute to nigrostriatal pathway degeneration and hastes progression of neurodegenerative diseases in humans.
The existence of ongoing inflammation process in PD is supported by findings of accumulation of cytokines, expecially IL-1β and NO metabolites, TNF alpha pathway activation and oxidative damage to molecules in the cerebrospinal fluid (CSF) of patients with PD mostly evident in post-mortem brain at autopsy.
Midbrain dopaminergic neurons display exquisite sensitivity to the death-inducing properties of cytokines such as IL-1β and, in addition, they live in a region reported toheve the highest density of microglia. Therefore if the latter become chronically activated, such as may occur with formation of fibrillar proteins' aggregates, the oxidative stress generated in this microenvironment might well tip the balance between life and death for nigral cells.
Because cytokines such as IL-1β are potent drivers of proximal cell death pathways, anti-inflammatory therapy could represent a promising neuroprotective intervention with the potential to delay or prevent onset of the disease. Several potential agents may open new avenues for the treatment of PD operating through different molecular mechanism such as enhancing the production of neurotropic factors or interfering with the apoptosis of these cells.
THE NEUROPROTECTIVE EFFECT OF MINOCYCLINE

Minocycline is a tetracycline antibiotic with a broader spectrum then the other members of the group. It is the most lipid-soluble of this class of antibiotics,giving it the greatest penetration into the brain across blood-brain barrier.
Commonly used against the infective agent Chlamydiophila Pneumniae, in vitro experiments and in various animal models of neurodegenerative disease, minocycline has demonstrated neuroprotective properties.
Parkinson' s disease has shown a particular beneficial response to minocycline in research studies. As an anti-inflammatory, the drug inhibits apoptosis in an indirect way downregulating pro-inflamatory cytokines' output aging on microglia and directly upregulating bcl-2 (an anti-apoptotic protein) syntesis. (Minocycline: neuroprotective mechanisms in Parkinson's disease,2004)
Minocycline most important effects are:
- Reducing neuroinflammation by blocking microglial activation in excitotoxicity that occurs after various brain injury and in consequence of formation of aggregates, as in Parkinson. Activated microglia release:
- The cytokine IL-1β
- Nitric Oxigen-metabolites, for the increased quantity of NO following the induction of i NOS
- Inhibition of p38-MAPK pathway
- Attenuation of apoptosis through two main mechanism:
- By blocking the release of caspase 1 and 3
- By upregulating the expression of the anti-apoptotic factor Bcl-2
It has been demonstrated that nigral cells' death occurs for glutamate exitotoxicity which is mediated through activation of NMDA-gated ion channels in several neurodegeneratice disease, Parkinson too. Mynocyclines blockes IL-1β converting enzyme and inducible nitric oxide synthase (iNOS) upregulation in animal models protecting neurons against NMDA excitotoxicity. NMDA treatment alone induces microglial proliferation which precedes dopaminergic cells death. Minocyclines inhibites this response to NMDA: it prevents the NMDA-induced proliferation of microglial cells and the release of IL-1β and NO mainly expressed by microglial coltures.
The cytokine IL-1β has emerged as the prime candidate underlying neuroinflammation that contribut to neurodegeneration. Its upregulation is the key of acute inflammatory processes in the Central Nervous System unfolding after a wide range of CNS insults, for example trauma and ischemia. But recent work has provided evidence for a harmful role of IL-1β in this setting driving substained neuroinflammation that contributes to the pathophysiology of neurodegenerative disease. An experiment with transgenic animals was conduced to confirm it: they demonstrate that sole over-expression of IL-1β was sufficient to drive a robust, chronic neuroinflammatory response.
Moreover it avoids the NMDA-related activation of p38 mitogen-activated protein kinase (MAPK) in microglia suggesting its potential effects for PD.

Activation of NMDA receptors (NMDA receptors) to a large extent mediates the glutamate toxicity (Glu-induced excitotoxicity) and neuronal death because of the toxic calcium influx which activates numerous enzyme including NOS: NO is able to further increase the excitotoxicity by enhancing glutamate release from presynaptic neurons and inhibiting astrocytes' glutamate transporters.
Several studies have suggested that the key players of excitotoxicity are parenchymal microglia that release a large variety of neurotoxine including NO and cytokines such as IL-1β further increasing microglia proliferation in a self-powered mechanism.
Statistically significative experiments show that NMDA-induced neuronal death involves activation of microglial cells and that minocycline prevents completely that and the microglial upregulated activity.
EXPERIMENTS AND DEMONSTRATION
A single-cell suspension taken from spinal cord (SC) is cultured and consists in 70%neuronal population, 25% astrocytes and 5% microglia and all express functional glutamate receptors. In a set of experiments, purified microglial cells are cultured on top of 5days-old SC and exposed at seventh day in vitro to 300 microM NMDA for 5minutes and analyzed 24 hours after the onset of exposure. This NMDA treatment reduces the number of survival neurons to 40-50%. To some cultures 20 nanoM minocycline is administered 30 minutes before the NMDA treatment.
Treatment with 20nanoM minocycline started before NMDA exposure is shown able to prevent neuronal death, confirmed in this cell culture model in that no apoptotic fragmentation is seen. The NMDA-induced neuronal death is associated with increase of NO metabolite production which is downregulated by 20nM of this anti-inflammatory compound.

An exposure to 300microM NMDA triggers microglial proliferation and activation: it is observed a 1.9-fold increase in the number of OX-42, a microglial marker of immunoreactive microglial cells in SC within 24 hours exposure.

Microglia release cytokines, expecially IL-1β and NO both riduced by antibiotical treatment and, if they are added on the top of SC cultures, they increase NMDA excitotoxicity which appear significantly, but not totally, inhibited by minocycline.
Minocycline prevents glutamate-induced apoptosis
Because MAPK have been reported to be involved in the robust gliosis in the SNpc which is accompanied by the induction of iNOS, the effect of the NMDA exposure was studied in SC cultures. The number of p38MAPK-positive microglia is significantly(130%) elevated after NMDA administration and is brought to basal levels again if 20nanoM of minocycline has been injected. Infact minocycline seems to inhibit iNOS expression. Researchers report that administration of this antibiotics potently blocks NO-induced neurotoxicity in vitro and, in vivo(mice model treated with MTPT,inducing PD-like symptoms), the degeneration of dopaminergic neurons. (Minocycline prevents nigrostriatal dopaminergic neurodegeneration in the MPTP model of Parkinson's disease,2001)

Note the marked reduction in TH-positive cell bodies after MTPT adminitration (compare a and b) and the protection exerted by minocycline (compare b and e)
Minocycline is known to inhibit p38 MAPK phosphorilation activity which is activated by a great number of cellular "stresses" and recently implicated in neuronal death induced by excitotoxicity. Moreover p38 MAPK mediates neuronal apoptosis induced by NO. Indeed a very recent report has implicated glial p38 MAPK in the neuroprotective action of minocycline. Data suggest that this type of tetracycline doesn't directly inhibit p38 MAPK activity but downregulates enzime activation through reducing its phosphorilation.
ATTENUATION OF APOPTOSIS

Apoptosis and the release of apoptosis-inducing toxines is a common mechanism of neurodegeneration. Minocycline reduces death of neurons: there an increasing evidence to suggest that the antiapoptotic effect is achieved through several actions at the level of mitochondrions. Minocycline stabilises mitochondria membranes and inhibits their permeability transition-mediated release of cytochrome c intothe cytosol, which is a potent stimulus for the activation of caspases 9 and 3 and the induction of apoptosis. The stabilisation of mitochondial membranes also reduces the release into the cytosol of others factors triggering both caspase-dependent and caspase-indipendent pathways leading to cell's death.
In neuronal cultures minocyclines upregulates the anti-apoptotic factor Bcl-2, which then accumulates in mitochondria to antagonise the pro-apoptotic Bax, Bak and Bid.
CONCLUSIONS

These data demonstrate that minocycline can effectively protect midbrain dopamine neurons through multiple beneficial mechanisms thanks to its high penetration into the brain, as it was observed in vitro experiments and in mice models. Neverthelss, nowaday any randomized trial clinial on a massive scale isn't conduced.
Can the cause be its low cost (approximately £4) not profitable enough for pharmaceutical companies?