Diphosphatidylglycerol or cardiolipin (CL) is a unique phospholipid with in essence a dimeric structure, having four acyl groups and potentially carrying two negative charges (the tetra-linoleoyl molecular species, important in heart mitochondria, is illustrated).

It is found almost exclusively in certain membranes of bacteria (plasma membrane and hydrogenosomes) and of mitochondria of eukaryotes (where it constitutes about 20% of the total lipid composition), i.e. those membranes whose function is to generate an electrochemical potential for substrate transport and ATP synthesis.
The trivial name "cardiolipin" is derived from the fact that it was first found in animal hearts, where it is especially abundant (10% of the phospholipids of bovine heart muscle) [Pangborn M., 1942]. However, it can be found in mitochondria of all animal tissues and indeed of the eukaryotic kingdom.
Until relatively recently, it was thought that CL was associated exclusively with the mitochondrial inner membrane. It is now know to occur in the mitochondrial outer membrane also if only at a level of about 4% (at sites connecting the outer membrane with the inner). The highly specific location of CL is used as an argument in favour of the hypothesis that mitochondria are derived from prokaryotes, which lived inside a eukaryotic progenitor cell in symbiosis. If this did indeed occur, the function of CL has changed during evolution, as mitochondria require a constant level of CL to function correctly, while prokaryotes only appear to require it in specific circumstances.
[Tian HF et al.: The evolution of cardiolipin biosynthesis and maturation pathways and its implications for the evolution of eukaryotes., 2012]
STRUCTURE
References for this part:
CL is a diphosphatidylglycerol lipid: two phosphatidylglycerols connect with a glycerol backbone in the center to form a dimeric structure. So it has four alkyl groups and can carry two negative charges.
As there are four distinct alkyl chains in CL, the potential for complexity of this molecule species is enormous. However, the compositions can be remarkably simple, very different from those of other phospholipids, and in animals they are resistant to dietary manipulation. Actually, in most animal tissues and in higher plants too, CL contains 18-carbon fatty alkyl chains, 80% of which is linoleic acid (18:2(n-6)). Although testis CL is an exception in that it contains mainly palmitic acid (16:0), while CL in the brain contains more fatty acids including arachidonic (20:4) and docosahexaenoic acids (22:6). Even if bacterial and yeast CL can differ in composition, a common feature of this lipid in a variety of very different organisms is a relatively simple fatty acid and molecular species composition, leading to a high degree of structural symmetry: it has been proposed that this 18:2 acyl chain configuration is an important structural requirement for the high affinity of CL to inner membrane proteins in mammalian mitochondria, the association with membrane proteins becomes looser upon double bond reduction.

Even with four identical acyl residues, CL has two chemically distinct phosphatidyl moieties, as two chiral centres exist, one in each outer glycerol group. In consequence, the two phosphate groups have different chemical environments, and they produce distinct 31 P-NMR resonances.

Each of the two phosphates in the molecule, designated 1'-phosphate and 3'-phosphate with respect to the central glycerol, can catch one proton. Ionizing one phosphate happens at a very different levels of acidity than ionizing both: pK1=2.8 and pK2>7.5. So, under normal physiological conditions, the molecule may carry only one negative charge.
The hydroxyl groups (–OH and –O-) on phosphate would form a stable intramolecular hydrogen bond with the central 2'-hydroxyl group. Molecular models of CL show that in aqueous dispersions its phosphates can form a tight bicyclic resonance structure with the central hydroxyl group. This resonance structure is especially stable when the molecule is symmetrical, so the four fatty acid constituents are identical.

Because of this unique structure, CL is able to form micellar, lamellar, and hexagonal states in aqueous dispersions, depending on pH and ionic strength. It is believed to exist mainly in a bilayer state in natural membranes, with its two phosphatidylglycerol moieties oriented perpendicular to the bilayer surface and the central glycerol group oriented at the water/membrane interface and parallel to it. However, there may remain a tendency of CL to form transient non-bilayer domains, which could have a profound influence on its function in vital cellular processes. The ability to form hexagonal phases is believed to be responsible for its location in the inner mitochondrial membrane at contact sites with the outer mitochondrial membrane.
As the head-group glycerol of CL is shared by two phosphatidate moieties, its mobility is severely restricted, reducing its capacity for either intra- or inter-molecular interactions with other phospholipid head-groups. Also, the secondary hydroxyl group of the central glycerol moiety is the sole source of hydrogen-bonding donor groups available for such interactions, so that bonding to other lipids is unlikely. It should be recognized that as the two phosphate groups are chirally distinct, there exist opportunities for differential activities or interactions of each under both chiral and achiral conditions.

MARKER
References for this part:
As the head group forms a compact bicycle structure, the head group area is quite small relative to the big tail region consist of 4 acyl chains. The fluorescent mitochondrial indicator, nonyl acridine orange (NAO), introduced in 1984, was found to target mitochondria by binding to CL [MeSH]. NAO has a very large head and small tail structure which can compensate with CL's structure, and arrange in a highly ordered way. Several studies were published utilizing NAO both as a quantitative mitochondrial indicator and an indicator of CL content in mitochondria: CL is termed the signature lipid of the mitochondrion.
However, it is found that NAO was influenced by membrane potential and/or the spatial arrangement of CL: so it's not proper to use NAO for CL or mitochondria quantitative studies in vivo.

BIOSYNTHESIS AND METABOLISM
Role of Cardiolipin in Mitochondrial Signaling Pathways, 2017
- The phospholipid cardiolipin (CL) is an essential constituent of mitochondrial membranes and plays a role in many mitochondrial processes, including respiration and energy conversion. Pathological changes in CL amount or species composition can have deleterious consequences for mitochondrial function and trigger the production of reactive oxygen species. Signaling networks monitor mitochondrial function and trigger an adequate cellular response. Here, we summarize the role of CL in cellular signaling pathways and focus on tissues with high-energy demand, like the heart. CL itself was recently identified as a precursor for the formation of lipid mediators. We highlight the concept of CL as a signaling platform. CL is exposed to the outer mitochondrial membrane upon mitochondrial stress and CL domains serve as a binding site in many cellular signaling events. During mitophagy, CL interacts with essential players of mitophagy like Beclin 1 and recruits the autophagic machinery by its interaction with LC3. Apoptotic signaling pathways require CL as a binding platform to recruit apoptotic factors such as tBid, Bax, caspase-8. CL required for the activation of the inflammasome and plays a role in inflammatory signaling. As changes in CL species composition has been observed in many diseases, the signaling pathways described here may play a general role in pathology.
References for this part:
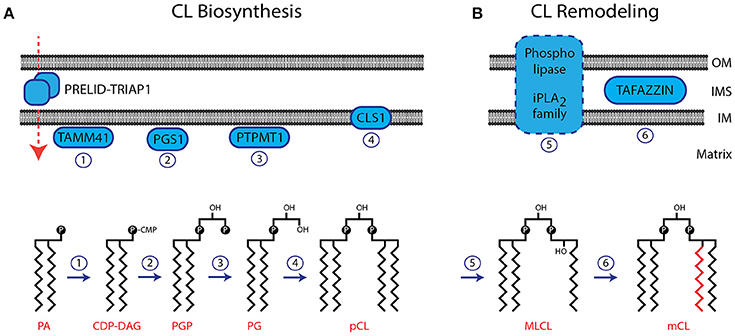
The biosynthetic pathway to CL is similar to that of some other phospholipids in that it passes through the common intermediates, phosphatidic acid and then cytidine diphosphate diacylglycerol. However, the final step is a unique reaction, which is very different in prokaryotes and eukaryotes. In the latter, it is the only phospholipid that is not synthesised on the cytosolic side of the endoplasmic reticulum but in the mitochondrion.
- Acylation of glycerol-3-phosphate by a glycerol-3-phosphate acyltransferase to form acylglycerol-3-phosphate, which can be acylated to form a phosphatidic acid (PA).
- CDP-DAG synthase (phosphatidate cytidylyltransferase) converts PA into cytidinediphosphate-diacylglycerol (CDP-DAG).
- PGP synthase converts CDP-DAG into phosphatidylglycerol phosphate (PGP), which is dephophorylated by PTPMT1 to form PG.
- Finally, a molecule of CDP-DAG is bind to PG to form one molecule of CL with elimination of cytidine monophosphate (CMP), catalyzed by the mitochondria-localized enzyme cardiolipin synthase (CLS).

In prokaryotes, CLS catalyses a transfer of the phosphatidyl moiety of one phosphatidylglycerol to the free 3'-hydroxyl group of another, with the elimination of one molecule of glycerol, via the action of two structurally related enzymes, which are part of the phospholipase D superfamily. In effect, transphosphatidylation occurs with one phosphatidylglycerol acting as a donor and the other an acceptor of a phosphatidyl moiety. The enzymes can operate in reverse under some physiological conditions to remove CL.
CARDIOLIPIN SYNTHASE
References for this part:
Cardiolipin synthase is a phosphatidyl transferase encoded by the CRLS1 gene, located on chromosome 20 (20p13-12.3). This gene is also called GCD10, due to homology between the deduced protein and S. cerevisiae Gcd10, an RNA-binding protein. It is located contrapodal to the MCM8 gene, which encodes for the highly conserved mini-chromosome maintenance proteins (MCM) that are essential for the initiation of eukaryotic genome replication.

In Homo sapiens there are two isoforms, which are transcriptional variants of the same gene:
- Isoform 1 is composed by 301 AA., and weights 32 462 Da
- Isoform 2 is composed by 202 AA., and weights 22 452 Da

The enzymes from all species examined in detail require certain divalent cations (Mg2+, Mn2+ or Co2+) together with a high pH (8 to 9). Cardiolipin synthase loses activity during purification, and the activity can be partially reconstituted by the addition of phospholipids (the most effective of which is phosphatidylethanolamine, which reactivates in a cooperative manner). CL reactivates hyperbolically at low concentrations but inhibits the enzyme at higher concentrations. In addition, CL shifts the sigmoidal reactivation curve of phosphatidylethanolamine toward lower concentrations. It is suggested that cardiolipin synthase requires interaction with several molecules of phosphatidylethanolamine and at least one molecule of CL for full enzymatic activity.
In rat liver, the cardiolipin synthase resides in the inner mitochondrial membrane as a multi-pass protein, while in yeast it is part of a large protein complex. The catalytic centre of cardiolipin synthase is exposed to the matrix side of the inner membrane.
As eukaryotic cardiolipin synthase is a mitochondrial enzyme and mitochondria are believed to be phylogenetic derivatives of ancient prokaryotes, it appears strange that there has been such a change in mechanism.
RE-MODELLING
The ultimate fatty acid composition of CL in eukaryotes is attained by re-modelling. This can be achieved by the coenzyme A (CoA)-dependent deacylation-reacylation cycle known as Lands cycle (after W.E.M. Lands), though it is now believed that the main route is via CoA-independent transacylation between different phospholipids in which the enzyme tafazzin plays a major part. This is necessary as the precursor phospholipids are very different in composition from that apparently required in the final product if it is to function correctly.
- Removal of a single acyl chain with formation of monolysocardiolipin (MLCL) by a calcium-independent phospholipase A2 (strong substrate preference for palmitoyl residues).
- In heart and yeast mitochondria, tafazzin transfers linoleate groups highly selectively from phosphatidylcholine to monolysocardiolipin, promoting molecular symmetry among the molecular species of fully acylated CL (and with formation of lysophosphatidylcholine).

The reaction does not require a coenzyme A ester as an intermediate, and it is reversible. This is believed to be the first CoA-independent phospholipid transacylase to have been identified, and it may be involved in re-modelling of other phospholipid classes.
The reaction is thus quite different from the cycle of acylation and deacylation involved in the remodelling of the more conventional phospholipids. Unlike the latter in which only position sn-2 is modified, all four positions in CL are affected. When the re-modelling reaction proceeds by the cyclic mechanism illustrated, only a trace level of either lyso-phosphatidylcholine or monolysocardiolipin need be present.
This mechanism may be an oversimplification in that two further enzymes have the ability to attach an acyl chain to monolysocardiolipin: monolysocardiolipin acyltransferase-1 (high specificity for linoleate) and acyl-CoA:lysocardiolipin acyltransferase-1.
In addition to the biogenesis of new CL, this remodelling process may be utilized to repair oxidatively damaged CL or to change the molecular form of existing CL to suit a specific mitochondrial function.
TAFAZZIN
References for this part:
Tafazzin is a protein encoded in humans by the gene TAZ, located on the X chromosome (Xq28). It is expressed at high levels in cardiac and skeletal muscle. Multiple transcript variants encoding different isoforms have been described. A long form and a short form of each of these isoforms is produced; the short form lacks a hydrophobic leader sequence and may exist as a cytoplasmic protein rather than being membrane-bound. RT-PCR revealed tissue-specific expression of several TAZ variants in leukocytes, fibroblasts, heart, and skeletal muscle.

This protein, functioning as phospholipid-lysophospholipid transacylase, was identified in 1996. Owing to the complex procedure required for the identification of tafazzin, the protein was named after "Tafazzi", a masochistic comic character in an Italian television show.
CATABOLISM
Catabolism of CL may occur by the action of phospholipase A2 to remove fatty acyl groups, possibly after oxidation as part of the process of apoptosis. There is also a specific mitochondrial phospholipase D, which hydrolyses CL to phosphatidic acid (and phosphatidylglycerol), promoting the fusion of mitochondria. This may be especially important under conditions of oxidative stress. The rate of hydrolysis by all phospholipases is significantly higher in the 3'-phosphatidyl moiety.
FUNCTIONS AND MECHANISMS
CL has some crucial functions in the inner membrane of mitochondria, where it interacts with a large number of mitochondrial proteins, thanks to its unique bicyclic structure. A change in pH and the presence of divalent cations can induce a structural change. CL shows a great variety of forms of aggregates. It is found that in the presence of Ca2+ or other divalent cations, CL can be induced to have a lamellar-to-hexagonal (LA-HII) phase transition. And it is believed to have a close connection with membrane fusion.
RESPIRATORY CHAIN
References for this part:
The respiratory chain consists of four enzymes (*NADH dehydrogenase, succinate dehydrogenase, cytochrome bc1 complex*, and cytochrome c oxidase) organized in large complexes (I to IV).
CL is an integral component of complex III, IV (2 molecules of CL required) and V (4 molecules of CL required) and is essential for the stability of the quaternary protein structure of the ADP-ATP carrier, which enables ATP and ADP to traverse the inner mitochondrial membrane. For this reason, it is an essential component of the interface between the complex and its membrane environment or between subunits within the complex. Removal of CL leads to break-up of the complex and loss of functionality.
Moreover, in relation to energy metabolism, it anchors two kinases: mitochondrial creatine kinase and nucleoside diphosphate kinase, to the inner and possibly the outer mitochondrial membranes where they come in contact. In this instance, and with the ADP-ATP- and phosphate-carrier proteins, it may facilitate the transport of solutes between the intra-membrane and matrix spaces of mitochondria.
Looking at the physical chemistry of the interaction between CL and enzymes, CL has a strong binding capacity for many structurally unrelated proteins, so its structure must be adapted to differing protein surfaces. This interaction has been studied intensively for the cytochrome bc1 complex, which couples electron transfer between ubiquinol and cytochrome c to the translocation of protons across the lipid bilayer. One CL molecule is bound close to the site of ubiquinone reduction and is believed to ensure the stability of the catalytic site as well as being involved in proton uptake. In general, the head group of CL and certain amino acid residues interact strongly via electrostatic forces, hydrogen bonds, and water molecules, while the acyl chains retain their flexibility and interact through van-der-Waals forces with the protein surface at a number of sites. However, we do not yet know why the highly specific fatty acid and molecular species compositions are necessary for these functions.
BUFFERING EFFECT
Thanks to its structure, CL function as a proton trap within the mitochondrial membranes during the oxidative phosphorylation process catalyzed by Complex IV, in which large quantities of protons are transferred from one side of the membrane to another side causing a large pH change. In this way it strictly localizes the proton pool and minimizes the changes in pH in the mitochondrial intermembrane space.

APOPTOSIS
References for this part:
CL is involved in the process of apoptosis through its interactions with a variety of death-inducing proteins, including cytochrome c. It is believed to act as a peroxidase, which reacts quite specifically with CL (but not with other more abundant phospholipids), causing oxidation and then hydrolysis of the product CL hydroperoxides, with the consequent conformation change. As a consequence, the cytochrome C is released into the inter-membrane space, while the oxidized CL is translocated to the outer mitochondrial membrane and participates in the formation of the mitochondrial permeability transition pore that facilitates egress of pro-apoptopic factors from mitochondria into the cytosol where they triggers apoptosis.
Moreover, during this process, CL is involved in the anchoring, translocation and embedding of caspase in the mitochondrial membrane, causing further release of apoptotic factors into the cytosol.
As a result, the cellular concentration of CL decreases rapidly while some mono-lyso-cardiolipin may accumulate.
OTHER FUNCTIONS
- CL is believed to be an important cofactor for cholesterol translocation from the outer to the inner mitochondrial membrane, and in steroidogenic tissues, it is a potent stimulator of steroidogenesis activating mitochondrial cholesterol side-chain cleavage.
- CL may also have a specific role in the import of proteins into mitochondria, and it can behave as a molecular chaperone to promote folding of mitochondrial proteins.
- It binds in a highly specific way to the DNA in eukaryotic chromatin: functional role in the regulation of gene expression.
- As a component of the plasma lipoproteins, it is believed to have an anti-coagulant function.
CARDIOLIPIN IN DISEASE
BARTH SYNDROME
References for this part:
Barth syndrome (BTHS) is an X-linked autosomal recessive disorder characterized by neutropenia, cardiomyopathy and growth retardation. It is a mitochondrial disorder caused by an inborn error of phospholipid metabolism. Prevalence is estimated at 1/454,000 and incidence at 1/140,000 (South-West England, South Wales) to 1/300,000-1/400,000 live births (USA). BTHS affects male patients.
The low concentration of CL in Barth fibroblasts is not the result of a low rate of synthesis, but it comes from an increased degradation. Under normal conditions, CL is degraded to mono-lyso-cardiolipin and then converted back into CL in order to exchange its fatty acids. In Barth fibroblasts, the deacylation–reacylation cycle seemed to be impaired because the incorporation of linoleic acid into CL was reduced. This observation in conjunction with the low CL level suggested a decline in the rate of reacylation relative to the rate of deacylation.
Tafazzin deficiency inhibits specifically the acyl remodeling of CL. So dysfunctional tafazzin is associated with defects in the assembly and/or function of the oxidative phosphorylation machinery, with an increased production of reactive oxygen species. The exact consequences for mitochondrial function remain to be established, but they may include deficiencies in mitochondrial energy coupling and/or in mitochondrial biogenesis.
Mitochondrial abnormalities in Barth syndrome compromise the development and function of certain tissues, such as muscle and heart, in which high energy turnover requires a strict structural organization of mitochondria both in terms of their morphology and their intracellular distribution. But the change of CL composition causes a consistent decrease of CL concentration also in platelets, granulocytes, lymphocytes, cultured lymphoblasts and fibroblasts.
In patient's peripheral blood nearly 50% of neutrophils contain "empty" cytoplasmic vacuoles, whereas there was an apparent maturation arrest of myeloid differentiation at the myelocyte stage. In addition, patients suffer from hypotonia, muscle weakness, undeveloped skeletal muscles, lack of stamina and distinctive exercise fatigue. Yet, some BTHS patients exhibit no cardiomyopathy manifestations, but suffer from neutropenia and frequent recurring episodes of bacterial infections.
ROLE OF CL IN TANGIER DISEASE
References for this part:
Tangier disease is a disorder characterized by very low blood plasma levels of high-density lipoprotein cholesterol, accumulation of cholesteryl esters in tissues and reduced cholesterol excretion in response to HDL apolipoproteins, increasing the risk for developing cardiovascular disease. Unlike Barth syndrome, it is caused by abnormal enhanced production of CL (three to five-fold), due to defects in the ATP binding cassette transporter 1, leading to cholesterol storage in TD cells.
This disorder has been first identified on the island of Tangier, on the coast of Virginia but people suffering from this disease are found all over the world. It is a rare autosomal recessive disease, fewer than 100 families are known, and it affects both genders equally.
Recent studies have shown that in TD fibroblasts the content of CL is increased. This may be linked to TD disease in two ways:
- The translocation of cellular cholesterol is closely related to translocation of CL and both processes are coordinately and impaired in TD.
- Enhanced synthesis of CL may reflect enhanced mitochondrial activity, as the result of an adaptive mechanism.
ANTIPHOSPHOLIPID SYNDROME
Antiphospholipid syndrome is an autoimmune, hypercoagulable state caused by antibodies against CL and apolipoprotein H. APS provokes thrombosis in both arteries and veins (mostly in vessels in which thrombosis is very uncommon, such as hepatic and renal veins) as well as pregnancy-related complications such as miscarriage, stillbirth, preterm delivery. In anti-cardiolipin-mediated autoimmune disease, there is a dependency on the apolipoprotein H (which binds CL) for recognition. [McNeil HP et al.: Anti-phospholipid antibodies are directed against a complex antigen that includes a lipid-binding inhibitor of coagulation: beta 2-glycoprotein I (apolipoprotein H)., 1990]

OTHER DISORDERS
Reductions in the concentrations of CL or changes in its composition in heart mitochondria have been implicated in many different human diseases states, including heart failure and diabetes, although it is not clear whether these effects are symptoms or the cause. For example, there is a dramatic decline in the content of myocardial CL at the onset of diabetes, and this is accompanied by extensive remodelling with the production of molecular species enriched in docosahexaenoic acid. "[Sparagna GC et al.: Loss of cardiac tetralinoleoyl cardiolipin in human and experimental heart failure., 2007]": http://www.ncbi.nlm.nih.gov/pubmed/17426348 ; [Han X et al.:Alterations in myocardial cardiolipin content and composition occur at the very earliest stages of diabetes: a shotgun lipidomics study., 2007].
Although oxidation of CL is part of the normal process of apoptosis, there is evidence that the proximity of this lipid to highly reactive oxygen species can lead to excessive peroxidation and oxidative stress, for example in the ischemic heart and skeletal muscle or during aging. Malfunctions of CL metabolism in brain mitochondria have been implicated in Alzheimer's disease and Parkinson's disease. [Ruggiero FM et al.: Lipid composition in synaptic and nonsynaptic mitochondria from rat brains and effect of aging., 1992] ; [Ellis CE et al.: Mitochondrial lipid abnormality and electron transport chain impairment in mice lacking alpha-synuclein., 2005].
DIAGNOSTIC USE
CL from a cow heart is used as an antigen in the Wassermann test for syphilis, based on complement-fixation. A specific form of CL is found in the membrane of Treponema palladium: syphilis non-specific antibodies (RPR or reagin) react with the lipid and the intensity of the reaction (1 to 4) indicates the severity of the condition. [MeSH]
Anti-cardiolipin antibodies can also be increased in numerous other conditions, including systemic lupus erythematosus, malaria and tuberculosis, so this test is not specific.
The presence of antibodies to CL in plasma of patients with various diseases in which tissue damage occurs is considered to be a danger signal to the immune system. T cells responsive to CL or oxidized CL may have a function in immune surveillance during infection and tissue injury, while antibodies to CL are used in diagnostic tests after unexplained venous or arterial thrombotic episodes or recurrent miscarriages.