INTRODUCTION
Hereditary hyperferritinemia cataract syndrome (HHCS) is a rare autosomal dominant genetic disease,first described in 1995 by two distinct reasearch groups, in Italy and France.
They reported two families in whom elevated serum L-ferritin concentration without iron overload, presenting with juvenile bilateral cataracts, was inherited as an autosomal dominant trait.

- inheriting a single mutant gene is sufficient for the disease to develop.
- even with only one of the two parents affected, the newborn has a 50% chance of inheriting the mutations that can then be passed to his/her own children.
EPIDEMIOLOGY
The prevalence of HHCS in different populations is unknown. A number of reports, mostly case reports, have previously been published. [3]
To this date, several affected families have been identified in different European countries, including Spain, and in other non-European regions, although the syndrome seems to be quite unusual. [6]
Nevertheless, this syndrome should be known by clinicians dealing with hyperferritinemia (i.e. hepatologists and hematologists) to avoid unnecessary and even dangerous phlebotomies.
CLINICAL FINDINGS
A distinguishing feature of HHCS is bilateral juvenile cataracts, usually during the second decade of life, which have an unusual morphology. They are described as “sunflower-type” morphology or “breadcrumb-like”. [2]
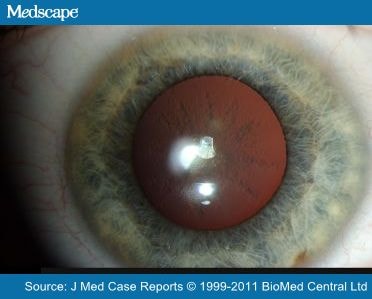
The opacities consist of abundant Lferritin protein. The precise mechanism by which this occurs is unclear. Lens opacities might be caused by a yet unknown interaction between L-ferritin and the lens proteins, or by a disturbed metabolism of L-ferritin within the lens. The high protein concentration in the lens, the slow turnover of mature lens fibers after formation and the surrounds of the avascular lens may also be involved in the interaction.
It is not yet determined whether cataracts in these patients is static or progressive.
No involvement of organs other than the eye has been reported in patients so far.
Ferritin levels in HHCS can exceed values over 6000 μg/L without any correlation to the severity of the affected lens.
ABOUT FERRITIN:
Ferritin is an ubiquitous intracellular protein that stores iron and releases it in a controlled fashion. The amount of ferritin reflects the amount of iron stored. The protein is produced by almost all living organisms, including algae, bacteria, higher plants, and animals. In humans, it acts as a buffer against iron deficiency and iron overload. [4]
Ferritin is a globular protein complex consisting of 24 protein subunits arranged in a particular way in order to create a cavity capable of storing up to 4500 Fe3+ ions as an inorganic complex.

Two different forms of ferritin exists in human body, varily composed by the three different subunities L, H, G:
1) Serum ferritin is a byproduct of intracellular ferritin synthesis . This form consists mainly of L subunits, which can also be glycosylated (G).
2) Intracellular ferritin : As an intracellular iron storage molecule,it is a heteropolymer composed of 24 H and L subunits, variously assembled.
Free iron is toxic to cells as it acts as a catalyst in the formation of free radicals from reactive oxygen species via the Fenton Reaction .

These free radicals can be quite harmful, particularly for cellular components such as lipids, proteins and nucleic acids.
- Lipid peroxidation , particularly of those lipids constituting plasma cell membranes or intracellular membranes (such as mitochondrial’s), is a common ROS-induced damage. Free radicals, when Oxigen is present, reacts with membrane’s lipid’s double bonds, generating lipidic peroxydes that can spread the damage throughout the membrane. In this case,the most dangerous ROS is •OH.
In erithrocytes, ROS can induce hemolysis.
- Protein Oxidation : free radicals can oxidize lateral aminoacidic groups, impairing protein function. They can promote cross-linkage formation such as disulfuric bond, causing mutations in protein’s structure or folding.
- DNA damage : DNA structure can be damaged or modified by free radicals, through the induction of mutations in azotate bases . This is how ROS can be responsible of cellular aging and cancer induction.
Hence vertebrates use an elaborate set of protective mechanisms to bind iron in various tissue compartments. Within cells, iron is stored in a protein complex as ferritin or hemosiderin.
Apoferritin (the form of ferritin not yet bound to iron) binds to free ferrous iron and stores it in the ferric state. As ferritin accumulates within cells of thereticuloendothelial system, protein aggregates are formed as hemosiderin. Iron in ferritin or hemosiderin can be extracted for release by the RE cells although hemosiderin is less readily available. Under steady state conditions, the serum ferritin level correlates with total body iron stores.
A normal ferritin blood level, referred to as the reference interval is determined by many testing laboratories. The ranges for ferritin can vary between laboratories but are usually between 30–300 ng/mL (=μg/L) for males, and 15–200 ng/mL (=μg/L) for females.
Serum ferritin concentration, though, is often fluctuating,not only in response to the amount of circulating/stored body iron, but also in response to stress or any ongoing inflammatory state (from infections to dismetabolism).
Variable hyperferritinemia levels are quite a common finding because ferritin is an acute phase reactant showing cytokine-inducible synthesis; these cytokines, in turn, are released in the course of inflammatory and tumor disorders (in fact, ferritin concentrations has been shown to increase drastically in the presence of an infection or cancer).
For example, endotoxin is an up-regulator of the gene coding for ferritin, thus causing the concentration of ferritin to rise.
Thus, the iron stores of the infected body are denied to the infective agent, impeding its metabolism.
All this implies that it can be considered an acute phase protein.
Ferritin synthesis is regulated by the availability of iron. An interaction between the IRP and the IRE of the FTL gene controls the translation of the L-ferritin gene. The IRE is a non-coding stem loop sequence located on the 5’UTR of the L-ferritin mRNA. In the presence of abundant cellular iron there is a structural change in the IRP, that prevents the IRP from binding to the IRE, and ferritin synthesis will proceed.
When there is a shortage of cellular iron, there is no relevant structural change and IRP binds to IRE and ferritin
translation is inhibited.
GENETICS AND PATOGENESIS
Hereditary hyperferritinemia-cataract syndrome is due to a mutation in the 5’ non-coding region of the ferritin L-chain gene, that maps at 19q 13.3-q 13.4 . This region, known as IRE (iron responsive element), interacts with two cytoplasmic proteins, known as iron regulatory proteins (IRPs). In iron deficiency status, IRPs acquire a high affinity for IRE, thus inhibiting the synthesis of L-ferritin chains. This interaction takes place on a loop structure formed by a 5 base-pair motif at an end of IRE.

Mutations in the loop or the adjacent stem structure prevent or decrease the interaction between IRE and IRPs, giving rise to a synthesis of ferritin L-chains that is not under the control of this regulatory system, and that is therefore independent from iron homeostasis.
At least 11 single nucleotide polymorphisms and 4 different deletions of variable length have been identified thus far . The closer to the IRE loop region a mutation is located, the more severe and earlier the syndrome results.
However, patients sharing the same mutation may show variability in clinical expression.
In this syndrome, hyperferritinemia results from the formation of L-chain monopolymers, or heteropolymers with a low number of H chains. The synthesis of Hchains is coded for by an independent gene that is located in chromosome 5 and that is normal in this syndrome.
The ability of the ferritin molecule to incorporate iron depends on its H-chain contents, and therefore ferritin molecules with excessive L chains in this syndrome can neither carry nor store iron. Because of this reason, no iron overload is present . The only known adverse consequence related to this genetic defect (i.e. cataracts) is due to the entrance and accumulation of L chains into the lens of affected subjects, which may reach values up to 15 or even 1500 above the normal range.
The early formation of cataracts is difficult to explain based only on a deposition of L chains, because they are soluble; it has been suggested that they would affect the solubility of other proteins or deteriorate the antioxidant defenses of the lens.
DIAGNOSIS
Clinicians should suspect it when treating any subject with early cataracts (particularly if bilateral and crumble-like) even more if they are familial, or in patients with very high levels of ferritinemia without evidence of iron overload (normal levels of serum iron and normal transferrin saturation) and normal haematological parameters. [1] , [5]
Genetical hemocromatosis should be excluded.
TREATMENT
There are no known consequences of the syndrome other than cataracts, there’s no evidence of iron overload and its proper diagnosis carries a favorable prognosis and eliminates the risk of unnecessary phlebotomies.
The only necessary treatment is surgical crystalline substitution.
BIBLIOGRAFY
[1] Hyperferritinemia without iron overload in patients with bilateral cataracts: a case series.2011
Hereditary hyperferritinemia cataract Syndrome: prevalence, lens morphology, spectrum of mutations, and clinical presentation.2003
Hereditary hyperferritinemia-cataract syndrome. Study of a new family in Spain.2004
Ferritin-Wikipedia
Hereditary hyperferritinaemia-cataract syndrome: a challenging diagnosis for the hepatogastroenterologist.2005
Hyperferritinaemia-cataract syndrome: worldwide mutations and phenotype of an increasingly diagnosed genetic disorder.2010