INTRODUCTION
Neuroleptic malignant syndrome (NMS) is a potentially fatal neurologic disorder and probably the most serious adverse reaction to neuroleptic medication. Firstly described by Delay and colleagues in 1960 in association with haloperidol, it was given the name of “syndrome malin de neuroleptics”.
EPIDEMIOLOGY
The estimated incidence rate of NMS is between 1% and 1.5% of patients treated with neuroleptics. Usually reported mortality rates vary from 11% to 38%. However, in recent studies they have fallen below 10% due to early recognition and improved management
CLINICAL FEATURES
NMS is characterized by the following symptoms: fever, muscle rigidity with cramps and tremor, autonomic nervous system instability (such as unstable blood pressure), alterations in mental status (catatonic state, agitation, delirium, or coma), seizures, tachycardia, tachypnea. Once symptoms appear, they may rapidly progress and reach peak intensity in as little as three days. These symptoms can last anywhere from eight hours to forty days. It is essential to recognize forms of incipient NMS (presenting with mental status changes, catatonia, rigidity, dysphagia or dysarthria), as any case can progress to a fulminant form that is more difficult to treat.
There are no laboratory findings specific for NMS, but elevated white blood cell count, low serum iron, metabolic acidosis, hypoxia, and increased serum CPK and catecholamines have been reported as frequent signs.
RISK FACTORS
NMS may be more likely to develop in patients with dehydration, agitation, low serum iron, underlying brain damage (Lewy body dementia and Parkinson) and catatonia.
Studies on relatives have suggested the presence of a genetic risk factor, as some patients may have genetic abnormalities in central dopamine systems that increase their susceptibility to NMS.
Anyway, one of the clearest risk factors in the development of NMS remains the course of drug therapy chosen to treat a condition. From 15 to 20% of patients who develop NMS have experienced a previous episode while taking antipsychotics, which is why taking a careful drug history is important. Furthermore, studies have demonstrated that rapid dose escalation during the treatment and cumulative antipsychotic dose may also trigger the development of NMS.
Demographically, it appears that males (80% less than 40 years), are at greatest risk for developing NMS, although it is unclear if the increased incidence is a result of greater neuroleptic use in men under forty. It has also been suggested that postpartum women may be at a greater risk for NMS.
Antipsychotic dose escalation as a trigger for Neuroleptic Malignant Syndrome, Langan J, Martin D, Shajahan P and Smith DJ, 2012
PATHOGENESIS
There are two traditional theories to explain NMS: the first is based on a neuroleptic induced alteration of central neuroregulatory mechanisms, the latter supposes an abnormal reaction of skeletal muscle.
First theory: central dopamine receptor blockade
Several important diseases of the nervous system are associated with dysfunctions of the dopamine system. The antipsychotic drugs that are frequently used to treat them have indeed a primary effect of attenuating dopamine activity.
Dopamine (alongside noradrenergic, serotoninergic and cholinergic pathways) actually plays a role in central thermoregulation in mammals: since neuroleptic drugs block dopamine receptor sites, the loss of heat dissipation associated with NMS may result from a blockade of hypothalamic dopamine sites. In addition, blockade of dopamine receptors in the corpus striatum seems to cause muscular rigidity, contributing to heat generation. The excess heat production, in association with a decrease in heat dissipation, produces hyperthermia, which is one of the main signs of the syndrome. However, it is unlikely that blockade of dopamine receptors in the hypothalamus and corpus striatum could completely explain all the signs of NMS.
Other studies have hypotized a genetically reduced function of dopamine receptors D2 and D3 as the main cause of the syndrome
Second theory: neuroleptic’s toxicity on muscle tissue
Release of calcium from the sarcoplasmic reticulum is increased with antipsychotic usage. This not only leads to heat generation, but (at higher calcium concentration) triggers activation of proteases, increased skeletal muscle cell contractility, mitochondrial dysfunction, and the production of reactive oxygen species, resulting in skeletal muscle cell death. All this can be related to the hyperthermia, breakdown of muscle, muscle rigidity, increase in creatine kinase levels observed in NMS.
A slightly different theory says that neuroleptics are not directly toxic to normal skeletal muscle, as we just said, but, basing on similarities between NMS and malignant hyperthermia, suggests that neuroleptic medications induce abnormal calcium availability in muscle cells of susceptible individuals and trigger muscle rigidity, rhabdomyolysis and hyperthermia. In a large proportion (50–70%) of cases, the propensity for malignant hyperthermia is due to a mutation of the ryanodine receptor 1 (RYR1), located on the sarcoplasmic reticulum. Some studies do actually support an association between NMS and the mutations in the ryanodine receptor gene reported with malignant hyperthermia.
Minor theories
Involvement of the serotoninergic system in NMS has been suggested. However, polymorphisms in the 5-HT1A and 5-HT2A receptor genes do not determine susceptibility to NMS.
A neuroimmunologic hypothesis is related to the idea that a fulminant acute phase response may account for the clinical and biochemical features of neuroleptic malignant syndrome. Leukocytosis, elevated muscle enzymes (particularly creatine phosphokinase), dramatic drop in serum iron levels are consistently observed in both neuroleptic malignant syndrome and the acute phase response. The objective of this theory is to demonstrate that the acute phase response is activated in this toxic drug reaction.

THE SYMPATHOADRENAL HYPERACTIVITY MODEL
As we just said, it is widely believed that dopamine D2 receptor antagonism causes hyperthermia in neuroleptic malignant syndrome by blocking heat-loss pathways in the anterior hypothalamus, by increasing heat production secondary to extrapyramidal rigidity, or both, but this model has been criticized as inadequate and lacking sufficient evidence. An alternative hypothesis is that neuroleptics are directly toxic to muscle tissue, as volatile anesthetics are in malignant hyperthermia, but neuroleptic malignant syndrome and malignant hyperthermia likely have distinct mechanisms. Neither model enables clinicians to identify specific patients at greater risk for neuroleptic malignant syndrome or to make reliable treatment choices.
In 1999 a pathophysiological model for neuroleptic malignant syndrome was developed. This model is related to a mechanism that induces disregulated sympathoadrenal hyperactivity and shows a greater explanatory power than the alternative hypotheses of hypothalamic dopamine antagonism (elevated set point) and direct myotoxicity (malignant hyperthermia variant). It is grounded on the fact that sympathetic nervous system mediates hypothalamic coordination of thermoregulatory activity and is a primary regulator of muscle tone and thermogenesis, augmenting both of these when stimulated. In addition, the sympathetic nervous system modulates all of the other end-organs that actually function abnormally in neuroleptic malignant syndrome. For instance, tachypnea and tachycardia in neuroleptic malignant syndrome reflect a hyperadrenergic state, while urinary incontinence is another clinical manifestation of autonomic dysfunction.
Cathecolamines and thermogenesis
There are two likely sources of excess thermogenesis: accelerated brown-adipose-tissue-like metabolism (uncoupled phosphorylation) and excessive catecholamine-induced Ca2+ release from the sarcoplasmic reticulum.
Brown adipose tissue is specialized to generate large amounts of heat rapidly, a process referred to as "nonshivering thermogenesis". Brown adipose tissue is rich in mitochondria containing uncoupling protein, which permits respiratory chain oxidation to liberate energy in the form of heat.
Persistent β-adrenergic stimulation induces brown adipose tissue hypertrophy and accelerates uncoupling protein gene transcription in rats, and although brown adipose tissue is probably not a major thermogenic tissue in man, it does persist throughout adulthood and can be reactivated following intense sympathetic stimulation.
The term for heat generated by the contractile apparatus in skeletal muscle is called “shivering thermogenesis”. Contraction is initiated when Ca2+ is released from its intracellular storage site in the sarcoplasmic reticulum and binds to contractile proteins (mainly troponin) in the sarcoplasm. Relaxation begins when intracellular Ca2+ concentration reaches a critical level, terminating further Ca2+ release and activating Ca2+ reuptake pumps in the sarcoplasmic reticulum membrane to move Ca2+ from sarcoplasm to sarcoplasmic reticulum against a steep gradient. Ca2+ release from the sarcoplasmic reticulum, Ca2+ binding to troponin, Ca2+ reuptake by the sarcoplasmic reticulum, and the oxidative chemical reactions of the postcontraction recovery phase all produce heat, and intracellular Ca2+ transport plays a role in normal thermogenesis. Intracellular free [Ca2+] regulates contraction-relaxation and agents that lead to sustained elevation of sarcoplasmic [Ca2+] can induce persistent contraction in the absence of an action potential.
Moreover, the sympathetic nervous system plays an integral role in normal muscle function. Force is potentiated by β-adrenoceptor agonists in mammalian skeletal muscle, and adrenoceptors on skeletal myocyte membranes are predominantly β2, indicating that epinephrine normally is directly involved in regulating skeletal muscle contraction. In skeletal and heart muscle fibers, β-adrenoceptor agonists promote phosphorylation of Ca2+ channels, increasing the probability of their being open. Epinephrine increases contraction strength by way of this altered Ca2+ influx, apparently by increasing the amount of Ca2+ released from the sarcoplasmic reticulum into sarcoplasm during activation (the so called phenomenon “calcium-induced calcium release”). Norepinephrine has similar effects. Thus, norepinephrine and epinephrine are both potent activators of skeletal muscle and exert this effect by altering intracellular [Ca2+].
Dopamine metabolism and involvement in thermogenesis

Dopamine (3,4-dihydroxyphenethylamine) is a neurotransmitter that, among other things, helps control the brain's reward and pleasure centres. Dopamine also helps regulate movement and emotional responses, and it enables us not only to see rewards, but to take action to move towards them.

First precursor of dopamine is tyrosine, which is taken up into dopamine nerve terminals via tyrosine transporter and converted into DOPA by the enzyme tyrosine hydroxylase. DOPA is then converted into dopamine by DOPA decarboxylase. After syntesis, dopamine is packaged into synaptic vesicles via the vesicular monoamine transporter (VMAT2) and stored until release into the synapse during neurotransmission.

Dopamine transporter (DAT) on the presynaptic terminal is responsible for clearing excess dopamine out of the synapse. There is also a presynaptic dopamine-2 autoreceptor, which regulates release of dopamine from the presynaptic neuron. In addition, there are several postsynaptic receptors (D1, D2, D3, D4 and D5), which constitute the primary binding site for virtually all antipsychotic agents.
We can find four major dopamine pathways in the brain: 1) The mesolimbic pathway (from the midbrain ventral tegmental area to the nucleus accumbens), involved in pleasurable sensations, the powerful euphoria in drugs abuse, as well as delusions and hallucinations of psychosis; 2) The mesocortical pathway (from the midbrain ventral tegmental area to the prefrontal cortex). It may have a role in mediating cognitive and affective symptoms (schizophrenia); 3) The nigrostriatal pathway (from the substantia nigra to the basal ganglia or striatum). It is part of the extrapyramidal nervous system and controls motor function and movement; 4) Tuberoinfundibular pathway (from the hypothalamus to the anterior pituitary gland), controlling prolactin secretion.
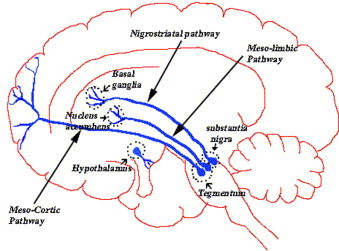
The hypothalamic dopaminergic pathways are widely believed to be involved in thermoregulation in mammals. This idea originates from the fact that sympathetic nervous system actually consists of potentially autonomous components whose functions are normally coordinated and integrated by the hypothalamus: the intrinsic capacity of the sympathetic nervous system for autonomous activity normally is suppressed by inhibitory regulatory inputs originating in the frontal cortex and mediated by hypothalamic nuclei. Thus, it seems that hypothalamus could integrate afferent thermosensory information and coordinate thermoeffector responses also by means of dopaminergic modulation of preganglionic sympathetic nervous system neurons.
Effects of neuroleptics use
As we said, the majority of commonly used antipsychotics act by blocking dopamine receptors. This leads directly to the idea that dopamine antagonists may interrupt tonic inhibitory modulation of sympathetic nervous system function, enhancing the effects of extreme psychic distress (altered frontal lobe function) on sympathoadrenal activity and causing profound disturbances of homeostasis. Disruption of these inhibitory inputs leads to dysregulated sympathetic nervous system hyperactivity with uncoordinated, excessive stimulation of end-organs by autonomous functional components of the sympathetic nervous system (vasomotor, sudomotor, inotropic, thermogenic, and others). Manifestations of dysregulated sympathetic nervous system hyperactivity include: increased muscle metabolism and tone (due to increased intracellular [Ca2+]), of which elevated creatine phosphokinase is one indicator; increased mitochondrial thermogenesis (caused by uncoupled oxidative phosphorylation); ineffective heat dissipation related to unregulated vasomotor and sudomotor activity; fluctuations in vasomotor tone leading to labile blood pressure, flushing, and pallor; granulocytosis and urinary incontinence. The combination of this effects produces the clinical syndrome of NMS.

Neuroleptic malignant syndrome, Adnet P, Lestavel P and Krivosic-Horber R, 2000
Relationship between functional dopamine D2 and D3 receptors gene polymorphisms and neuroleptic malignant syndrome, Mihara K, Kondo T, Suzuki A et Al, 2003
Neuroleptic malignant syndrome: a neuroimmunologic hypothesis, Anglin RE, Rosebush PI and Mazurek MF, 2010
Sympathoadrenal Hyperactivity and the Etiology of Neuroleptic Malignant Syndrome, Gurrera RJ, 1999
ANTIPSYCHOTICS

At the molecular level, NMS, as we said, is probably caused by a sudden, marked reduction in dopamine activity. Especially individuals using haloperidol or chlorpromazine are reported to be at greatest risk. It is commonly thought that NMS incidence declined using second generation (or “atypical”) agents as clozapine instead of conventional (“typical”) antipsychotics. They both act on dopaminergic pathways, but second generation neuroleptics are less likely to cause important side effects. Above all they seem to reduce the risk of extrapyramidal motor control disabilities (rigidity and tremors).
However, providing proof that atypical agents are less likely to cause NMS is actually difficult. Why?
1) NMS is uncommon; its incidence in psychiatric patients treated with conventional antipsychotics is very low. To demonstrate reduced NMS incidence with atypicals, a large sample of patients would be required to reach statistical significance.
2) As doctors have used lower doses of conventional agent (which reduces the risk of NMS) any beneficial impact from atypicals has become more difficult to detect.
3) Reports of NMS frequency with atypicals may be inflated by bias in publishing adverse events with newer versus older agents.
4) Patients switched to atypicals may represent a high-risk group that is intolerant or resistant to conventional neuroleptics.
Neuroleptic malignant syndrome: Still a risk, but which patients may be in danger?, Caroff SN, 2003
TREATMENT
NMS is a medical emergency, and can lead to death if untreated. The following measures are usually applied:
1) First step is to stop neuroleptic drugs.
2) Treat the hyperthermia aggressively, such as with cooling blankets or ice packs to the axillae and groin.
3) Supportive care in an intensive care unit capable of circulatory and ventilatory support is crucial.
4) The correct pharmacological treatment is still unclear. Dantrolene has been used when needed to reduce muscle rigidity, and more recently dopamine pathway medications such as bromocriptine have shown benefit. Amantadine is another treatment option due to its dopaminergic and anticholinergic effects. Benzodiazepines may be used to control agitation.
5) Highly elevated blood myoglobin levels can result in kidney damage, therefore aggressive intravenous hydration with diuresis may be required.
Memory impairment is a consistent feature of recovery from NMS. It is usually temporary, though in some cases, may become persistent.
CONCLUSIONS
NMS is still not totally understood and its underlying pathophysiology remains uncertain. However, there is substantial evidence to support the hypothesis that dysregulated sympathetic nervous system hyperactivity may be responsible for most, if not all, features of neuroleptic malignant syndrome. A predisposition to more extreme sympathetic nervous system activation and/or dysfunction in response to emotional or psychological stress may constitute a trait vulnerability for neuroleptic malignant syndrome, which, when coupled with state variables such as acute psychic distress or dopamine receptor antagonism, produces the clinical syndrome of NMS.
This is the pathway that research shall follow in the future in order to obtain a comprehensive and definite explanation for this complex syndrome. In the meantime, a judicial choice of neuroleptic medication and careful observation of patients can reduce the incidence, morbidity, and mortality of the syndrome.