INTRODUCTION
Pendred syndrome is a genetic disorder leading to a congenital bilateral sensorineural hearing loss, with occasional goitre and hypothyroidism. It is named after doctor Vaughan Pendred, an English doctor, described the condition in an Irish family in 1896. It accounts for 7.5% of all cases of congenital deafness.

CLINICAL DESCRIPTION
Children who are born with Pendred syndrome may begin to lose their hearing at birth or by the time they are three years old. Usually, their hearing will worsen over time. The loss of hearing often happens suddenly, although sometimes some hearing will be restored afterwards. Eventually, some children with Pendred syndrome become totally deaf. After all there are also problems with normal language development, because cortical areas of hearing and speaking are largely overlapped. Childhood hearing loss has many causes. In the United States, researchers believe that 50–60 percent of cases are due to genetic causes, and 40–50 percent of cases result from environmental causes. Health care professionals use different clues, such as when the hearing loss begins and whether there are anatomical differences in the ears, to help determine whether a child has Pendred syndrome or some other type of progressive deafness.
Pendred syndrome also can affect the vestibular system, which controls balance. Some people with Pendred syndrome will show vestibular weakness when their balance is tested. However, the brain is very good at making up for a weak vestibular system, and most children and adults with Pendred syndrome don't have a problem with their balance or have difficulty doing routine tasks. Some babies with Pendred syndrome may start walking later than other babies, however.
hearing impairment
Classically, hearing loss is bilateral, severe to profund and congenital; in some instance it may be later in onset and progressive. High-resolution CT scans was used to assess temporal bone anatomy and it shows absence of the upper turn of the cochlea (diagnosed when the interscalar septum cannot be seen between the upper and middle turns) and deficiency of the modiolus. This condition it is aso known as Mondini dysplasia. This type of malformation has been described as a flattened cochlea with development only of the basal coil. Instead of two and one-half turns in the bony cochlea, there may be only one and one-half turns, with the middle and apical turns occupying a common space or cloaca.
CT-Scans of Cochlear Implant Patients with Characteristics of Pendred Syndrome-Vol. 32, No. 7, 2013
vestibular disfunction
Objective evidence of vestibular dysfunction can be demonstrated in 66% of individuals with PDS and ranges from mild unilateral canal paresis to gross bilateral absence of function. Also EVA (enlarged vestibular aqueduct) could be found in patients affected.

thyroid disfunction
Pendred syndrome can also involve thyroid hormones' equilibrium. It can make the thyroid gland grow larger. An enlarged thyroid gland is called a goitre. The thyroid is a small, butterfly-shaped gland in the front of the neck, just above the collarbones. The thyroid plays a major role in how the body uses energy from food. In children, the thyroid is important for normal growth and development. Children with Pendred syndrome, however, rarely have problems growing and developing properly even if their thyroid is affected. Their levels of thyroid hormones are usually normal. People with Pendred syndrome are significantly more likely than the general population to develop a goiter in their lifetime, although not everyone who has Pendred syndrome gets a goiter.

These data suggested that some other causes, especially environmental (geographical, dietary) ones, should take part in the onset of thyroid dysfunction.
In the kidney we later analyse how much genetic and environmental components cooperate in the onset of metabolic alkalosis. (see case report below).
DIAGNOSIS
Antenatal diagnosis is checked with DNA test of fetal cells or with the analysis of chorionic villus.
Clynical diagnosis is effected with:
- tonal audiometry, measuring both aereal an bone transmission in right and left ear. Results (showed in the figure), often indicate binaural deafness.

- RMN or TAC of the ear
- functioning of the thyroid gland with ecography
- functioning of renal excretion with ecography
- perclorate potassium test
- genetic research:Therefore genetics studies showed Mutations in three known genes that account for approximately half of PDS/DFNB4 cases: SLC26A4 (~50% of affected individuals), FOXI1 (<1% of affected individuals), and KCNJ10 (<1% of affected individuals), suggesting further genetic heterogeneity.
THE GENE
This condition is inherited in an autosomal recessive pattern, which means both copies of the gene in each cell have mutations. The parents of an individual with an autosomal recessive condition each carry one copy of the mutated gene, but they typically do not show signs and symptoms of the condition. Biallelic mutations in SLC26A4 are known to cause PDS. Sequence analysis of SLC26A4 identifies disease-causing mutations in approximately 50% of affected individuals from either simplex or multiplex families. These persons are often compound hetero-zygotes for disease-causing variants in SLC26A4 although not infrequently only a single variant is detected. The SLC26A4 gene is located on chromosome 7q21-31 and contains 21 exons with an open reading frame of 2343 bp.

Correlation between genotype and phenotype in patients with bi-allelic SLC26A4 mutations-2013 Sep 5
Digenic inheritance: rare reports of digenic inheritance, in which an affected individual has double heterozygosity with a heterozygous mutation in SLC26A4 and a heterozygous mutation in FOXI1 or double heterozygosity with a heterozygous mutation in SLC26A4 and a heterozygous mutation in KCNJ10 , are included in Table 1 and discussed in more detail in Molecular genetics .This type of digenic inheritance has not been supported by other studies, suggesting that mutations in FOXI1 and KCNJ10 are rare in this condition.
Table1
Molecular Genetics
FROM THE GENE TO THE PROTEIN
The SLC26A4 gene, which was cloned in 1997, encodes for a protein named pendrin, so that the mutation of the gene leads to a lack in the expression of this protein. Pendrin belongs to the SLC26A family, which includes several anion transporters, as well as the motor protein prestin, that is expressed in outer hair cells;this family, also known as anion multifunction exchanger. Slc26a4 solute carrier family 26, member 4.
The transcription of the gene is provided by some transcriptional factors whose activity is increased by massive exposure to inflammatory interleuchins ( IL4 and IL13).These are two of many small glycoproteins acting as signaling molecules in the humoral immune system. Once bound to their membrane-located receptor, the Janus kinase/signal transducer and activator of transcription ( JAK/STAT ) signal transduction cascade is activated. This cascade involves activation of the receptor by phosphorylation, followed by the recruitment, binding, phosphorylation and activation of STAT monomers. Currently, seven STAT isoforms have been identified, and STAT6 is the isoform preferentially activated in the presence of IL-4. Once phosphorylated on tyrosine residue 641, STAT6 monomers dimerize, at which point they are able to translocate from the cytosol into the nucleus. The STAT6 homodimers then preferentially bind to conserved DNA sequences referred to as interferon- γ activated sequences (GAS) motifs. So in the presence of the T-helper type-2 (TH2) cytokines, interleukin IL-4 and IL-13, pendrin expression is rapidly and robustly increased in a larger variety of tissues.
The Human Pendrin Promoter Contains two N4 GAS Motifs with Different Functional Relevance-Epub 2013 Dec 18.
PENDRIN
LOCALIZATION
Pendrin is located at the apical membrane three main types of cells in the organism: follicular cell in thyroid, endocochlear hair cell in the inner ear and in tubular cell in the kidney, where it is involved in the mechanisms of control of the acid-base equilibrium, exchanging anions such as HCO3- and Cl-.

MOLECULAR STRUCTURE
Pendrin is a a highly hydrophobic membrane glycoprotein composed of 780 aminoacids.
PENDRIN AMINOACIDS PERCENTAGE

It contains three putative extracellular asparagine-glycosylation sites. Pendrin usually appears as a single protein band with a molecular mass of 110–115 kDa when isolated from human thyroid membranes. Pendrin is proposed to have 12 transmembrane domains with both amine and carboxylic-termini located inside the cytosol. Like other members of the SLC26A family, pendrin contains a so-called STAS (sulfate transporter and antisigma factor antagonist) domain. The exact function of this domain has not been elucidated. Recent studies, however, suggest that the STAS domain can interact with the regulatory domain of CFTR (cystic fibrosis transmembrane conductance regulator) in certain epithelial cells.

MAIN FUNCTIONS
THYROID GLAND
In follicular cells of the gland, the function of this protein is that to provide the transport of the anion Iodine from the cell into the follicular lumen. This ion, after oxidation by TPO, is essential for the synthesis of the thyroid hormones T3 and T4; infact the absence of pendrin also lead to goitre an hypothyrodism, due to a lack in the production of thyroid hormones. Goiter usually develops during childhood. There is, however, a substantial variation within and between families and different geographic regions. Nutritional iodide intake appears to play an important role as a modifier of the thyroidal phenotype. Under conditions of high iodide intake, most individuals have no or only a mild enlargement of the thyroid. If the nutritional iodide is scarce, patients with Pendred syndrome not only develop goitre but may also present with mild or severe hypothyroidism.

Identification of PENDRIN (SLC26A4) Mutations in Patients With Congenital Hypothyroidism and Apparent Thyroid Dysgenesis-Epub 2013 Dec 20.
THE EAR
But the most relevant, and also interesting role of pendrin is its crucial function in granting a normal hearing. In the inner ear its function is different from that in the thyroid: Pendrin doesn’t transport iodine but it becomes antitransport of chlorine and bicarbonate; This anions exchange is not important for hormones synthesis but to maintain/regulate electrolytic equilibrium in endolimphatic structures such as cochlea and vestibular aqueduct. It is probably enroled in the active exchange of HCO3- an Cl- between endolymph and extra-cellular fluid and thereby in the regulation of the endolymphatic pH (that muts remain near plasma pH values). Previous studies shows Cochlear cell types with diffuse cytoplasmic staining for H+ATPase and a basolaterally localized Cl-/HCO3- exchanger ( AE2 ) including inner hair cells, root cells and a subset of supporting cells in the organ of Corti. Hair cells of the utricle, saccule and cristae ampullaris also expressed both H+ATPase and the Cl-/HCO3- exchanger, but immunostaining for the H+ATPase was less intense and less polarized than in the cochlea. These immunocytochemical results supported a role for the H+ATPase and Cl-/HCO3- exchanger in the regulation of endolymphatic pH and suggest that certain cells (including strial marginal cells and epithelial cells of the endolymphatic sac) may be specialized for this regulation. Later studies identify this in an apical exchanger ( pendrin ) and its specific localization in cochlea, in the apical membrane of these cells (where it provide the re-adsorption of HCO3- that returns iside the cells, while Cl- goes outside in the endolymph):
- spiral ganglion
- spiral ligament (near vascular stria)
- claudius cells
- deiters cells
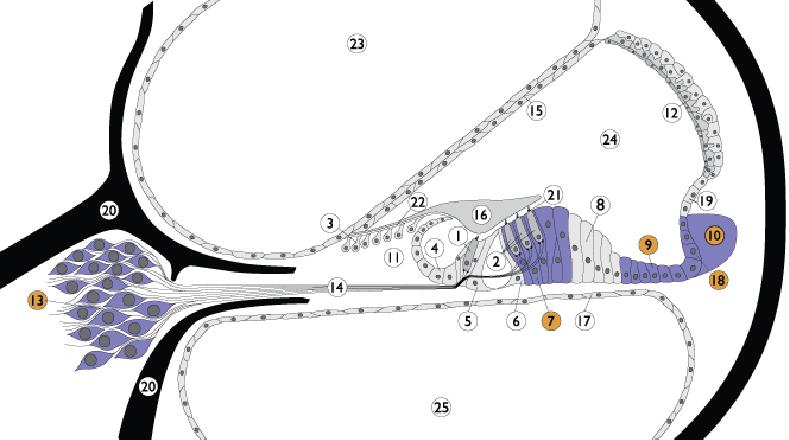
1. Inner hair cell; 2. Outer hair cell; 3. Interdental cells; 4. Inner sulcus cells; 5. Inner pillar cells; 6. Outer pillar cells; 7. Deiter’s cells; 8. Hensen cells; 9. Claudius cells; 10. Spiral ligament; 11. Spiral limbus; 12. Stria vascularis; 13. Spiral ganglion; 14. Auditory nerve; 15. Reissner’s membrane; 16. Tectorial membrane; 17. Basilar membrane; 18. External sulcus cells; 19. Spiral promi-nence; 20. Bony spiral lamina; 21. Reticular lamina; 22. Between IDC and TM; 23. Scala vestibuli; 24. Scala media
The effective role of pendrin in deafness, however remains unclear. Nowadays, with the development of a mouse with a targeted disruption of the Slc26a4, a new insight has revealed an additional role of pendrin in the inner ear: it is responsible for the re-absorption of the endolymph. Infact the absence of this protein in areas of the endolymphatic comartments known to play this role, leads to a profound prenatal endolymphatic hydrops and destruction of many of the epithelial cells surrounding the scala media. The precise mechanisms underlying endolymph reabsorption in the inner ear are not yet known; these studies, however, provide some of the groundwork for allowing the future delineation of these processes.
THE KIDNEY
an effective correlation between PDS and metabolic alkalosis?
New interesting insights into the role of pendrin in regulating acid-base homeostasis are still to be approved but a new case report could be able to show unexpected results.
a case report
A 46-year-old Caucasian female was admitted to hospital after being found on the floor of her home, confused and with rigid limbs. The medical history elicited from her family included childhood-onset sensorineural hearing loss and mild hypothyroidism that had been diagnosed at the time of cochlear implantation 1 year earlier (to afford an EVA and single cavity cochlea). There was a long history of alcohol excess. Her only medication was levothyroxine 50 μg daily. Initial examination revealed a small goitre, a sinus tachycardia (120 beats/min) and reduced oxygen saturation (91% on air). Tone in all limbs was increased with bilateral carpal spasms and rightward tonic deviation of the neck.
Initial arterial blood gas measurements indicated a severe metabolic alkalosis with compensatory hypoventilation: pH 7.59, pO2 7.08 kPa, pCO2 6.39 kPa, bicarbonate (HCO3) 45 mmol/l and base excess +20.4 (normal ranges 7.36–7.44; 9.3–13.3 kPa; 4.5–6.0 kPa; 22–26 mmol/l and −2 to +2 respectively). Venous biochemistry showed severe hypokalaemia and hypochloraemia. In addition, her deranged liver function tests, low platelets, severe hypomagnesaemia and raised mean cell volume (MCV) were consistent with chronic excessive alcohol intake. Inflammatory markers and lactate level were elevated, suggestive of infection. Her thyroid function tests were indicative of either under-replacement or non-compliance.
Venous biochemical and haematological parameters at presentation
Central venous access was obtained for fluid and electrolyte resuscitation, with close monitoring of fluid balance. She desaturated and developed a severe respiratory acidosis 24 h after admission, culminating in respiratory arrest that required intubation and ventilation. In the ICU, she received a continuous infusion (20–30 mmol/h) of potassium with hourly electrolyte measurement, but also required several infusions of magnesium (each 20 mmol/2 h), together with i.v. boluses of calcium gluconate. A ventricular fibrillation (VF) cardiac arrest responded to DC cardioversion and i.v. amiodarone 36 h post-admission, but she experienced several further VF arrests despite improvements in her venous K+ and Mg+ levels.
Her electrolyte balance stabilized by day 4, but her recovery after extubation was complicated by a chest infection requiring antibiotic treatment. Her potassium and calcium levels eventually normalized without ongoing replacement. Her elevated random blood glucose level normalized without treatment, suggesting that this was a stress response. Oral magnesium supplementation on discharge (day 37) was discontinued 4 weeks later, after which her electrolyte balance remained normal. Her blood pressure was 130/80 mmHg when she was well.
But in addition this woman was a heavy drinker. This can explain itself several problems of the woman in :
- muscular:the women presented rigid limbs, according to the fact that alcohol assumption determined a lower food-assumption so less K+ less Fe2+ and most of all less vitamin D. This is an important regulator of mitochondrial respiration that, if it is appropriate (with a NADH/NAD ratio of 40 per cent) leads to inositol production. This molecule is a really important second messenger controlling protein synthesis. In the muscle, its deficiency in this woman was the cause of her muscular/articular pains.
- perseverant VF and tachycardia: alcohol played an important role because it increases the conversion of T4 in T3, via activating the deiodinase in the liver. T3 is the most active of the thyroid hormones and its action over the heart was in contrast with the general condition of hypothyroidism due to pendred syndrome.
In the kidney the absence of a functional isoform of pendrin was probably the main cause of her renal insufficiency (but not the only only one). In fact in normal people the pendrin-expressing β-intercalated cells in the collecting duct are bicarbonate secreting,thus, under ambient conditions, also individuals with Pendred syndrome can maintain normal acid–base homeostasis. But the diet of this woman was inappropriate: the excess of alcohol lead to ipokalemia, this lead to a major uptake of H+ ions in the cells and their excretion in the lumen, and this lead to an increasing in pH. So this two causes together determined a life-threatening alkalosis: alcohol abuse and mutation in SLC26A4.
Only a single previous case of metabolic alkalosis in the context of Pendred syndrome has been reported. In that report, a child with the disorder developed severe metabolic alkalosis and hypokalaemia, but in the context of commencement of thiazide diuretic therapy for endolymphatic hydrops, presumably being unable to compensate for drug-induced renal chloride and potassium losses, leading to alkalosis. In summary, this patient's unusual clinical presentation highlights that in the kidney, pendrin does play a part in maintaining acid–base homeostasis in humans, with its absence and consequent failure of renal bicarbonate excretion leading to potentially life-threatening metabolic alkalosis in the context of inter-current illness.
Life-threatening metabolic alkalosis in Pendred syndrome. Eur J Endocrinol. 2011 July
THERAPY
From the clinical therapy point of view no treatment is available. It’s impossible, nowadays, to recover from the mutation of SLC26A4 gene, although research is improving a lot in this direction. By studying mice, scientists are gaining a greater understanding of how an abnormal SLC26A4 gene affects the form and function of different parts of the body. For example, by studying the inner ears of mice with SLC26A4 mutations, scientists now realize that the enlarged vestibular aqueduct associated with Pendred syndrome is not caused by a sudden stop in the normal development of the ear. Studies such as this are important because they help scientists rule out some causes of a disorder while helping to identify areas needing more research. Eventually, researchers are hopeful that these studies will lead to therapies that can target the basic causes of the condition.
It is possible, however, to successfully contrast symptoms so that the patient could live a completely normal life. In matter of the hypoacusia, treatment is represented by rehabilitating with acoustic prosthesis and cochlear implant. Also language support could be useful: children with Pendred syndrome should start early treatment to gain communication skills, such as learning sign language or cued speech or learning to use a hearing aid. Most people with Pendred syndrome will have hearing loss significant enough to be considered eligible for a cochlear implant. A cochlear implant is an electronic device that is surgically inserted into the cochlea. While a cochlear implant doesn't restore or create normal hearing, it bypasses injured areas of the ear to provide a sense of hearing in the brain. Children as well as adults are eligible to receive an implant. Regarding the thyroid disorder, while present, a simple pharmacological chronic substitution therapy with L-tyroxin is usually sufficient.