WHAT IS MYOSITIS OSSIFICANS?
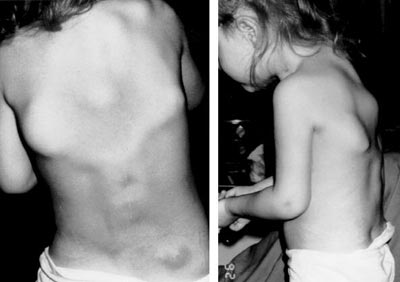 Myositis ossificans is a disorder in which muscle tissue and connective tissue, such as tendons and ligaments, are replaced by bone ( ossified ) , forming bone outside the skeleton ( extra-skeletal or heterotopic bone ) that constrains movement. Extra-skeletal bone formation causes progressive loss of mobility as the joints become affected; most ( i.e. 80%) ossifications arise in the thigh or arm(in nonhereditary type they are caused by a premature return to activity after an injury)and other sites include intercostal spaces, erector spinae, pectoralis muscles, glutei and the chest. (Myositis Ossificans,Wikipedia)
Myositis ossificans is a disorder in which muscle tissue and connective tissue, such as tendons and ligaments, are replaced by bone ( ossified ) , forming bone outside the skeleton ( extra-skeletal or heterotopic bone ) that constrains movement. Extra-skeletal bone formation causes progressive loss of mobility as the joints become affected; most ( i.e. 80%) ossifications arise in the thigh or arm(in nonhereditary type they are caused by a premature return to activity after an injury)and other sites include intercostal spaces, erector spinae, pectoralis muscles, glutei and the chest. (Myositis Ossificans,Wikipedia)
When the chewing muscles are involved, the inability to fully open the mouth may cause difficulty in speaking and eating and patients may experience malnutrition due to their eating problems. They may also have breathing difficulties as a result of extra bone formation around the rib cage that restricts expansion of the lungs.
(Fibrodysplasia ossificans progressiva,2007)
CLASSIFICATION
Myositis ossificans comprises two syndromes characterized by heterotopic ossification ( calcification ) of muscle:
- In the first, and by far most common type, nonhereditary myositis ossificans , calcifications occur at the site of injured muscle, most commonly in the arms or in the quadriceps of the thighs.
The term myositis ossificans traumatica is sometimes used when the condition is due to trauma.
- The second condition, myositis ossificans progressiva (also referred to as "fibrodysplasia ossificans progressiva", FOP ) is an inherited affliction, with autosomal dominant pattern , in which the ossification can occur without injury, and typically grows in a predictable pattern. Although this disorder can be passed to offspring by those afflicted with FOP, there are cases in which it can be attributed to a spontaneous genetic mutation of a gene; this type of disease is called nonhereditary even though it isn’t due to a trauma.
We are going to analyze the main features of FOP, fibrodysplasia ossificans progressive.
( Myositis Ossificans,Wikipedia)
WHICH GENE IS RELATED TO FIBRODYSPLASIA OSSIFICANS PROGRESSIVA?
 Mutations in the ACVR1 gene cause fibrodysplasia ossificans progressiva.
Mutations in the ACVR1 gene cause fibrodysplasia ossificans progressiva.
The official name of this gene is “ activin A receptor, type I ” and provides instructions for making the activin receptor type I protein, which is a member of a protein family called bone morphogenetic protein (BMP) type I receptors . BMP receptors are a family of transmembrane serine/threonine kinases receptors that span the cell membrane so that one end of the protein remains inside the cell and the other end projects from the outer surface of the cell. This arrangement allows receptors to receive signals from outside the cell and transmit them inside to affect cell development and function. The ligands of these receptors are members of the TGF beta superfamily . TGF-betas and activins transduce their signals through the formation of heteromeric complexes with 2 different types of serine (threonine) kinase receptors: type I receptors of about 50-55 kD and type II receptors of about 70-80 kD. Type II receptors bind ligands in the absence of type I receptors, but they require their respective type I receptors for signaling, whereas type I receptors require their respective type II receptors for ligand binding.
( BMP1A receptor, Wikipedia)
WHERE IS THE ACVR1 GENE LOCATED?
Cytogenetic Location : 2q23-q24
Molecular Location on chromosome 2 : base pairs 157,736,445 to 157,875,861

The ACVR1 gene is located on the long (q) arm of chromosome 2 between positions 23 and 24.
Activin receptor type I is found in many tissues of the body including skeletal muscle and cartilage. It helps to control the growth and development of the bones and muscles, including the gradual replacement of cartilage by bone (ossification). It is normally activated at appropriate times by ligands and the activation may occur when these ligands, such as BMPs, bind to the receptor or to other proteins with which it forms a complex. A protein called FKBP12 can inhibit activin receptor type I by binding to the receptor and preventing inappropriate (leaky) activation in the absence of ligand. This process occurs in normal skeletal maturation from birth to young adulthood.
All individuals with a definite diagnosis of FOP have a heterozygous missense point mutation in which the protein aminoacid histidine is substituted for the amino acid arginine at position 206 of the ACVR1 protein (written as Arg206His or R206H ). Researchers believe that, under certain conditions, this mutation may change the shape of the receptor and may disrupt the binding of an inhibitor protein, such as FKBP12, or interfere with other mechanisms that control activation. As a result, the receptor may be constantly activated ( constitutive activation ), even in the absence of ligands and the constitutive activation of the receptor causes overgrowth of bone and cartilage and fusion of joints, resulting in the signs and symptoms of fibrodysplasia ossificans progressiva .
( ACVR1 gene,2007)
HOW DO PEOPLE INHERIT FIBRODYSPLASIA OSSIFICANS PROGRESSIVA?
This condition is inherited in an autosomal dominant pattern, which means one copy of the altered gene in each cell is sufficient to cause the disorder.
Most cases of fibrodysplasia ossificans progressiva result from new mutations in the gene. These cases occur in people with no history of the disorder in their family.
(Fibrodysplasia Ossificans Progressiva,2007)
CLASSIC CLINICAL FEATURES OF FOP
Two clinical features define classic FOP:
- malformations of the great toes (this abnormality of the big toes is a characteristic feature that helps to distinguish this disorder from other bone and muscle problems)

- progressive heterotopic endochondral ossification (HEO) .
Individuals with FOP appear normal at birth except for characteristic malformations of the great toes which are present in all classically affected individuals. During the first decade of life, most children with FOP develop episodic, painful inflammatory soft tissue swellings. While some of them regress spontaneously, most transform soft connective tissues( including aponeuroses, fascia, ligaments, tendons, and skeletal muscles) into mature heterotopic bone. Ribbons, sheets, and plates of heterotopic bone replace skeletal muscles and connective tissues through a process of endochondral ossification that leads to an armament-like encasement of bone and permanent immobility.

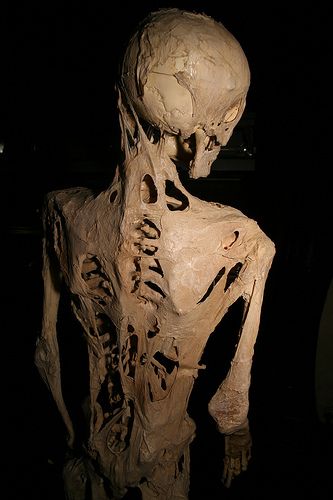
While malformation of the great toes is characteristic of FOP, other developmental anomalies are frequently observed such as stiffness of the neck , that is an early finding in most patients and can precede the appearance of HEO at that site. Characteristic anomalies of the cervical spine include large posterior elements, tall narrow vertebral bodies, and fusion of the facet joints between C2 and C7. Other skeletal anomalies associated with FOP include short malformed thumbs, clinodactyly (is a medical term describing a bend or curvature of the fifth fingers,the "little fingers" toward the adjacent fourth fingers (Clinodactyly), short broad femoral necks , and proximal medial tibial osteochondromas ( is a type of benign tumor that consists of cartilage and bone (Osteochondroma ) .

Minor trauma such as intramuscular immunizations, mandibular blocks for dental work, muscle fatigue, blunt muscle trauma from bumps, bruises, falls, or influenza-like viral illnesses can trigger painful new flare-ups(swellings) of FOP leading to progressive heterotopic ossification.
Heterotopic ossification in FOP progresses in characteristic anatomic and temporal patterns that mimic the patterns of normal embryonic skeletal formation and several skeletal muscles, including the diaphragm, tongue, and extra-ocular muscles, cardiac muscle and smooth muscle are spared from heterotopic ossification .
Bone formation in FOP is episodic, but disability is cumulative. Most patients with FOP are confined to a wheelchair by the third decade of life, and require lifelong assistance in performing activities of daily living. Severe weight loss may result following ankylosis of the jaw. Pneumonia or right-sided heart failure may complicate rigid fixation of the chest wall. The severe disability of FOP results in low reproductive fitness , and fewer than 10 multigenerational families are known worldwide. The median age of survival is approximately 40 years , and death often results from complications of thoracic insufficiency syndrome .
( The Medical Management of Fibrodysplasia Ossificans Progressiva: Current Treatment Considerations,2011)
HISTOPATHOLOGY OF FOP LESIONS

The histopathology of FOP lesions has been well-described.
Early FOP lesions contain an intense infiltration of macrophages, mast cells, and lymphocytes . The precise role of these cells in the evolution of FOP flare-ups is unknown although focal inflammation from any cause is a known trigger of disease activity. Subsequent migration of inflammatory cells into affected muscle, indeed, precedes widespread death of skeletal muscle. Following a rapid and destructive inflammatory stage, there is an intense fibroproliferative phase associated with robust angiogenesis and neovascularity . As lesions mature, fibroproliferative tissue undergoes an avascular condensation into cartilage followed by a revascularization stage with osteogenesis in a characteristic process of HEO. The resultant new ossicle of heterotopic bone appears histologically normal with mature lamellar bone and often contains marrow elements.
HOW COMMON IS FIBRODYSPLASIA OSSIFICANS PROGRESSIVA?
Fibrodysplasia ossificans progressiva is a very rare disorder, believed to occur in approximately 1 in 2 million people worldwide and shows no geographic, ethnic, racial, or gender preference.
(Fibrodysplasia Ossificans Progressiva,2007)
DIAGNOSIS
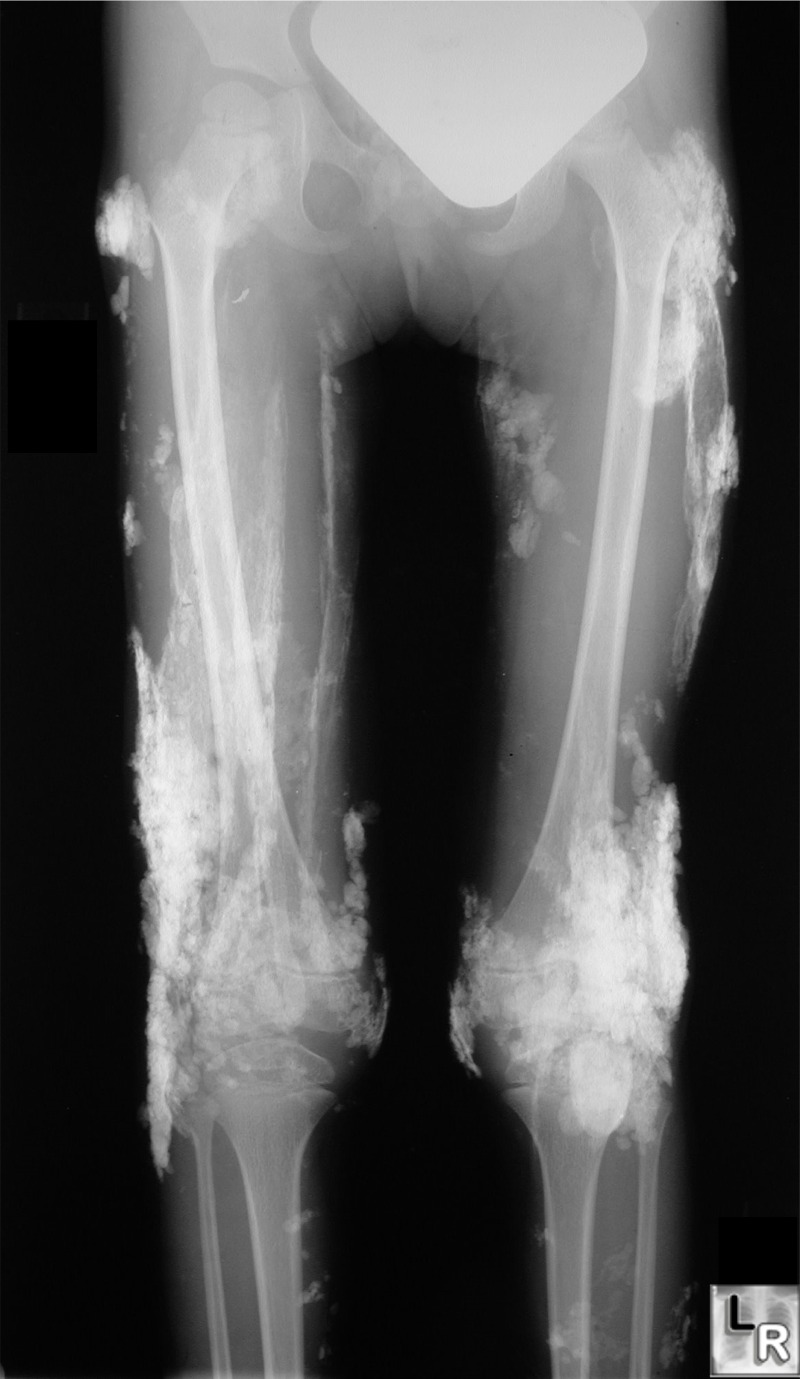
Radiographic findings suggest normal modeling and remodeling of the heterotopic skeleton. Nevertheless, multiple subtle skeletal abnormalities and joint malformations are often seen in individuals with FOP. However, the definitive diagnosis of FOP can be made by simple clinical evaluation that associates rapidly appearing soft tissue lesions with malformations of the great toes.
( The Medical Management of Fibrodysplasia Ossificans Progressiva: Current Treatment Considerations)
Definitive genetic testing of FOP is now available and can confirm a diagnosis of FOP prior to the appearance of heterotopic ossification. The test is realized with sequence analysis of the entire coding region using bi-directional Sanger Sequence Analysis (Test for Fibrodysplasia Ossificans Progressiva)
and the sensitivity of DNA sequencing is over 99% for the detection of nucleotide base changes, small deletions and insertions in the regions analyzed (Test for Fibrodysplasia Ossificans Progressiva).
Clinicians should be aware of the early diagnostic signs of FOP, which are congenital malformation of the great toes and episodic soft tissue swelling even before the appearance of heterotopic ossification. This awareness should prompt genetic consultation and testing (if appropriate) and the institution of assiduous precautions to prevent injury and iatrogenic harm.
CHALLENGES OF THERAPEUTIC ASSESSMENT IN FOP
Flare-ups of FOP are sporadic and unpredictable, and there is great individual variability in the rate of disease progression. Several large studies on the natural history of FOP have confirmed that it is impossible to predict the occurrence, duration or severity of an FOP flare-up , although characteristic anatomic patterning has been described. The rarity of FOP and the unpredictable nature of the condition make it extremely difficult to assess any therapeutic intervention, a fact recognized as early as 1918 by Julius Rosenstirn (Rosenstirn, 1918):
“The disease was attacked with all sorts of remedies and alternatives for faulty metabolism; every one of them with more or less marked success observed solely by its original author but pronounced a complete failure by every other follower. In many cases, the symptoms of the disease disappear often spontaneously, so the therapeutic effect (of any treatment) should not be unreservedly endorsed.”
These words ring true today as they did when they were written nearly a century ago.
Since the chirurgical therapy led to no results(attempts to surgically remove heterotopic bone, indeed, provoked explosive and painful new bone growth )the ultimate treatment of FOP will likely be based on integrated knowledge of the cellular and molecular pathophysiology of the condition.
- Corticosteroids
The rational use of corticosteroids early in the course of an FOP flare-up is based primarily on its potent anti-inflammatory effects and on emerging knowledge of the importance of inflammatory triggers in FOP flare-ups.
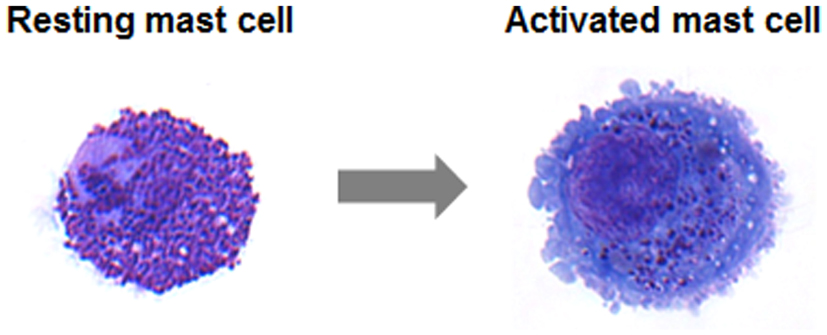
As most patients and families know all too well, a lesion may appear within hours and can reach an alarming size literally overnight. The sudden appearance and rapid spread of an FOP lesion suggests involvement of an armada of inflammatory mediators along with an abnormal connective tissue wound response, and points to a potential role for inflammatory mast cells and their mediators in the extension of the disease process.
- Cyclo-oxygenase 2 Inhibitors & NSAIDs
The body produces two types of prostaglandins : “physiological” prostaglandins and “inflammatory” prostaglandins. Physiological prostaglandins are normally produced in many of the body’s tissues and serve to protect organs, such as the stomach, from metabolic injury. Inflammatory prostaglandins are produced in response to injury, and play a major role in the inflammatory response to tissue injury and repair. Traditional NSAIDs (non-steroidal anti-inflammatory drugs)such as aspirin, ibuprofen and indomethacin inhibit the formation of both the physiological and inflammatory prostaglandins. The selective cyclo-oxygenase-2 (cox-2) inhibitors primarily inhibit the inflammatory prostaglandins and leave most, but not all, of the physiological prostaglandins relatively intact.
- Muscle Relaxants
Early FOP flare-ups are associated with intense mast cell, macrophage, and lymphocytic infiltration into skeletal muscle and are often accompanied by intense inflammatory changes within regions of locally damaged or necrotic skeletal muscle. Areas of relatively healthy skeletal muscle, bordering the lesion, are thus subject to metabolic changes that would lead to muscle spasm and fiber shortening . The short-term use of muscle relaxants may help to decrease muscle spasm and maintain more functional activity even in the setting of an evolving FOP lesion. This is especially true for painful flare-ups involving the major muscle groups of the back and limbs.
- Bone Marrow Transplantation
A recently published study documented the role of hematopoietic stem cells in FOP . Bone marrow derived stem cells have been implicated in the ectopic bone formation of FOP. The replacement of these stem cells by bone marrow transplantation has been suggested as a possible cure for FOP. However, the definitive contribution of bone marrow derived stem cells to the formation of heterotopic bone has remained obscure.
- Avandia (Rosiglitazone)
A recent case report by Gatti et al from the University of Verona approaches the problem from a different perspective. The report claims that rosiglitazone (Avandia), an anti-inflammatory and anti-diabetic agent that alters the fate of marrow stromal cells, “is associated with major clinical improvements in a patient with fibrodysplasia ossificans progressiva (FOP).” (Gatti et al., 2010) 
- Retinoic Acid Receptor Agonists
As far back as the 1980s, retinoids, used for the treatment of acne, were known to cause skeletal birth defects if taken during pregnancy because they interfere with the formation of the cartilaginous scaffold on which the embryonic skeleton is built. The idea of using retinoids to treat FOP flare-ups was simple and elegant: if retinoids caused birth defects by disrupting the formation of the cartilaginous scaffold of the normal skeleton, perhaps they might retard the formation of the cartilaginous scaffold of the heterotopic or second skeleton of FOP.
- Targeting the gene ACVR1: Definitive Targets for Therapy
The identification of the recurrent heterozygous missense point mutation, that causes FOP in all classically affected individuals, provides a specific pharmaceutical target and a rational point of intervention in a critical signaling pathway. The discovery of the FOP gene identifies ACVR1 as a susceptible 30 pharmaceutical target for the treatment of FOP. Plausible therapeutic strategies to inhibiting BMP signaling in FOP include:
- inhibitory RNA technology
- monoclonal antibodies directed against ACVR1
- orally available small molecule selective signal transduction inhibitors (STIs) of ACVR1
( The Medical Management of Fibrodysplasia Ossificans Progressiva: Current Treatment Considerations,2011)
HOW TO OBTAIN INFORMATION ABOUT FOP?
Obtaining tissue samples for extensive lab study is extremely difficult because injuries and surgeries can trigger rapid bone formation in FOP patients. One new approach is to use human stem cells, called induced pluripotent stem cells (iPS cells) , that we can create from donors with genetic diseases. The method consists in insert the genes OCT4, SOX2, KLF4, and C-MYC using retroviruses or episomes in patient’s cells (it is called " Yamanaka method " ).

Researchers at the University of California – San Francisco (UCSF) and Kyoto University used skin cells of patients with FOP to derive iPS cells and these were their results:
- ACVR1 (R206H) mutation is not sufficient to induce mineralization of fibroblasts
FOP patients show dramatic heterotopic bone formation in their soft tissues, yet do not ossify their skin. Having observed that, they asked if the ACVR1 R206H mutation could induce mineralization in primary cells that do not normally form bone. Human dermal fibroblasts (HDFs) from control donors and FOP donors were cultured, genotyped and propagated in HDF maintenance medium. For our mineralization assays, we used a culture medium that did not contain any exogenous BMPs that might mask the effects of the ACVR1 R206H mutation. This medium still promoted mineralization in primary human MSCs when compared with standard non-mineralizing MSC maintenance medium.
However, FOP HDFs showed no differences in mineralization activity when compared to HDFs cultured in non-mineralizing HDF maintenance medium, even with an additional 6 days of culture. Thus, the ACVR1 R206H mutation was not sufficient to induce spontaneous mineralization in HDFs in vitro.
- Human iPS cells with the ACVR1 mutation are pluripotent
Isolating primary tissues from FOP patients is extremely difficult since injuries, including surgical procedures, induce bone formation flares. iPS cells derived from a small skin biopsy or excess surgical material provide a potential way to create a continuous supply of diseased tissues in vitro for experimentation.
They first created iPS cell lines from banked FOP skin fibroblasts. Retroviruses withOCT4, SOX2, KLF4, and C-MYC were transfected into two control and two FOP fibroblast lines. Two control iPS cell lines and four FOP iPS lines were characterized in detail and showed the expected genotypes. Although low levels of exogenous KLF4 and SOX2 were detected in some lines, the iPS cell lines still expressed genetic markers of pluripotency and could form all three germ layers in teratomas;indeed, large amounts of cartilage were evident in several of the teratomas derived from FOP iPS cells.

- FOP iPS cells show increased chondrogenesis
The researcers tested if the FOP mutation might affect chondrogenesis in a directed in vitro chondrogenesis assay. Quantitative PCR showed elevated mRNA levels of SOX9 and COL2a1 (markers of immature chondrocytes) and COMP (marker of mature chondrocytes) in the pellets.
- Integration-free FOP iPS cells also show increased mineralization
They were created integration-free iPS cells to test if retroviral transgenes confound the possibility to detect the effects of the ACVR1 R206H mutation. We used integration-free episomal vectors to introduce the iPS cell transformation factors SOX2, KLF4, OCT4, L-MYC, LIN28, and p53 shRNA into control and FOP dermal fibroblasts. They obtained a large number of FOP iPS cell colonies with morphology consistent with human ES cells.
The iPS cell strategy provides a key towards dissecting the cellular and molecular mechanisms of human skeletal disease pathogenesis in FOP. This new FOP model shows that the mutated ACVR1 gene can affect different steps of bone formation, each of which may be potential targets for limiting or stopping the progression of the disease. Human iPS cells will also be a valuable tool that reflects the diversity of patients we see in the clinic since patients, and thus their iPS cells, show clinical variability, genetic background effects, and epigenetic influences. Their studies demonstrate the creation of iPS cells from patients with FOP, identify that ACVR1 may have a new role in regulating mineralization activity, provide a proof for the development of human iPS cells as disease models for studying human skeletal diseases and for guiding future drug development .
(Induced pluripotent stem cells from patients with human fibrodysplasia ossificans progressiva show increased mineralization and cartilage formation,2011)