Introduction
MLD, also known as Arylsulfatase A deficiency, is a Lysosomal Storage Disease which is commonly listed in the family of leukodystrophies as well as among the sphingolipidoses as it affects the metabolism of sphingolipids.
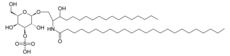
Metachromatic leukodystrophy (MLD) is a severe neuro-metabolic disorder caused by a deficiency in the lysosomal enzyme Arylsulfatase A which catalyzes degradation of 3-O-sulfogalactosyl-ceramide, an essential and abundant component of myelin.
Wikipedia
MLD is also considered a Neurodegenerative Disease because the Storage of Sulfatides (sulphated glycosphingolipids, especially sulfogalactosylceramides or sulfogalactocerebrosides) in the oligodendrocyte and Schwann cells causes progressive demyelination in the central and peripheral nervous systems. Due to the pervasive nature of this disease, metabolic alterations in MLD may go beyond sulfatide accumulation and involve other metabolites.
Their incidence ranges between 0.5 and 1/50 000 and prevalence is estimated at 1 case in 625 000.
MLD is autosomal recessive and results from an inability to metabolise cerebroside sulphate. In most cases, the deficient enzyme is Arylsulfatase-A, the gene for which has been located on chromosome 22 (22q). In the late infantile form, the activity of arylsulfatase A is very low to non-existent. In the juvenile form, enzymatic deficiency and sulfatiduria are also present but less accentuated than in the infantile form, while in the adult form, residual enzymatic activity is found. Arylsulfatase A is activated by Saposin B (Sap B), a non-enzymatic proteinaceous cofactor. In rare cases, mutations have been found in the gene coding for this activator involved in the enzymatic hydrolysis of the lipids, called SAP-B, located on chromosome 10 (10q21-22). When the Arylsulfatase A enzyme level is normal but the sulfatides are still high - meaning that they are not being broken down because the enzyme is not activated the resulting disease is Saposin B Deficiency. The enzyme that is present is not "enabled" to a normal level of efficiency and can't break down the sulfatides which results in all of the same MLD symptoms and progression.
A recent study contended sulfatide is not completely responsible for MLD because it is nontoxic. It has been suggested lysosulfatide, sulfatide which has had its acyl group removed, plays a role because of its cytotoxic properties in vitro.
Google
Google
NCBI


Wikipedia
Disease characteristics
Arylsulfatase A deficiency (also known as metachromatic leukodystrophy or MLD) is characterized by three clinical subtypes: late-infantile MLD (50%-60% of cases); juvenile MLD (20%-30% of cases); and adult MLD (15%-20% of cases). Age of onset within a family is usually similar. The disease course may be from three to ten or more years in the late-infantile form and up to 20 years or more in the juvenile and adult forms.
• Late-infantile MLD. Onset is between ages one and two years. Typical presenting findings include weakness, hypotonia, clumsiness, frequent falls, toe walking, and slurred speech. Later signs include inability to stand, difficulty with speech, deterioration of mental function, increased muscle tone, pain in the arms and legs, generalized or partial seizures, compromised vision and hearing, and peripheral neuropathy. In the final stages children have tonic spasms, decerebrate posturing, and general unawareness of their surroundings.
• Juvenile MLD. Onset is between age four years and sexual maturity (age 12-14 years). Initial manifestations include decline in school performance and emergence of behavioral problems, followed by clumsiness, gait problems, slurred speech, incontinence, and bizarre behaviors. Seizures may occur. Progression is similar to but slower than the late-infantile form.
• Adult MLD. Onset occurs after sexual maturity, sometimes not until the fourth or fifth decade. Initial signs can include problems in school or job performance, personality changes, alcohol or drug abuse, poor money management, and emotional lability; in others, neurologic symptoms (weakness and loss of coordination progressing to spasticity and incontinence) or seizures predominate initially. Peripheral neuropathy is common. Disease course is variable, with periods of stability interspersed with periods of decline, and may extend over two to three decades. The final stage is similar to that for the earlier-onset forms.

Diagnosis/testing
MLD is suspected in individuals with progressive neurologic dysfunction and MRI evidence of a leukodystrophy. MLD is suggested by arylsulfatase A (ARSA) enzyme activity in leukocytes that is less than 10% of normal controls; however, assay of ARSA enzymatic activity cannot distinguish between MLD and ARSA pseudodeficiency, in which ARSA enzyme activity that is 5% to 20% of normal controls does not cause MLD. Thus, the diagnosis of MLD must be confirmed by one or more of the following additional tests: molecular genetic testing of ARSA (the only gene in which mutations are known to cause arylsulfatase A deficiency), urinary excretion of sulfatides, and/or finding of metachromatic lipid deposits in nervous system tissue.
Clinical Diagnosis
Arylsulfatase A deficiency is suspected in individuals with the following
- Progressive neurologic dysfunction. Presenting signs may be behavioral or motor. Symptoms can occur at any age beyond one year and follow a period of normal development
- MRI evidence of a leukodystrophy
- Diffuse symmetric abnormalities of periventricular myelin with hyperintensities on T2-weighted images. Initial posterior involvement is observed in most late- infantile cases with subcortical U-fibers and cerebellar white matter spared. As the disease progresses, MRI abnormalities become more pronounced in a rostral-to-caudal progression; cerebral atrophy develops.
- Anterior lesions may be more common initially in individuals with later onset.
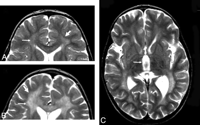
Scoring details of the metachromatic leukodystrophy (MLD) MR imaging severity score. Differences between scoring faint (1 point) versus dense appearance (2 points) are shown. A, Periventricular and central involvement in the frontal white matter is categorized as faint (1 point,thin arrow), whereas myelination is preserved in the subcortical U-fibers (0 points, thick arrow). B, Periventricular and central involvement in the frontal white matter is categorized as dense (2 points, thin arrow), and there are areas that lack subcortical U-fiber myelination (2 points, thick arrow). C, Inner atrophy as measured in the third ventricle (arrow).
AJNR

Midline of the corpus callosum in advanced stages of MLD. T2 lesion hyperintensities in the midline are reduced compared with the adjacent supratentorial white matter lesion signal intensity (arrow).
AJNR
Read more: Metachromatic leukodystrophy: a scoring system for brain MR imaging observations
NCBI
Read more: Lysosomal storage diseases : Diagnostic confirmation and management of presymptomatic individuals
Nature
Testing
Arylsulfatase A enzyme activity
• Arylsulfatase A (ARSA) enzyme deficiency. ARSA enzyme activity in leukocytes is less than 10% of normal controls using the usual Baum type assay in which other arylsulfatases are incompletely blocked
• ARSA enzyme pseudodeficiency. ARSA enzyme activity in leukocytes is 5% to 20% of normal controls. Pseudodeficiency is difficult to distinguish from true ARSA enzyme deficiency by biochemical testing alone.
Because assay of ARSA enzymatic activity cannot distinguish between MLD and ARSA pseudodeficiency, the diagnosis of MLD is confirmed by one or more of the following additional tests:
1. Molecular genetic testing of ARSA
2. Urinary excretion of specialized compounds.
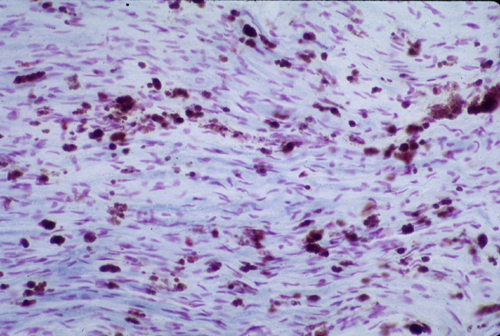
3. Metachromatic lipid deposits in a nerve or brain biopsy. Sulfatides interact strongly with certain positively charged dyes used to stain tissues, resulting in a shift in the color of the stained tissue termed metachromasia. When frozen tissue sections are treated with acidified cresyl violet (Hirsch-Peiffer stain), sulfatide-rich storage deposits stain a golden brown. The finding of metachromatic lipid deposits in nervous system tissue is pathognomonic for MLD.
Molecular Genetic Testing: Gene ARSA is the only gene in which mutations are known to cause arylsulfatase A deficiency. Three classes of ARSA alleles resulting in low ARSA enzyme activity need to be distinguished:
1. ARSA alleles with a mutation that causes MLD (ARSA-MLD alleles)
2. ARSA alleles with sequence variants resulting in pseudodeficiency (ARSA-PD)
3. Alleles with two ARSA sequence variants on the same chromosome (cis configuration).
Expert-revieves
Management
There is currently no treatment or cure for MLD. Children with advanced juvenile or adult onset and late infantile patients displaying symptoms receive treatment limited to pain and symptom management.
Treatment of manifestations : Treatment of seizures using antiepileptic drugs in standard protocols; treatment of contractures with muscle relaxants; physical therapy and an enriched environment to maximize intellect, neuromuscular function, and mobility; family support to enable parents and/or caregivers to anticipate decisions on walking aids, wheelchairs, feeding tubes, and other changing care needs.
Prevention of primary manifestations : Hematopoietic stem cell transplantation (HSCT) or bone marrow transplantation (BMT), the only therapies for primary central nervous system manifestations, remain controversial because of their substantial risk and uncertain long-term effects. The best outcomes are observed when HSCT or BMT is performed before symptoms occur. These therapies may slow down progression of the disease or stop its progression in the central nervous system.
Prevention of secondary complications :Physical therapy to prevent joint contractures by maintaining joint mobility facilitates nursing care in the later stages of the disorder. ; routine health care maintenance.
Genetic counseling
MLD is inherited in an autosomal recessive manner. At conception, each sib of anaffected individual has a 25% chance of being affected, a 50% chance of being an asymptomatic carrier, and a 25% chance of being unaffected and not a carrier. This is important in clarifying the genetic status of at-risk relatives. Carrier testing of at-risk family members and prenatal diagnosis for pregnancies at increased risk are possible if both disease-causing ARSA mutations have been identified in an affected family member
Therapies Under Investigation
Attempts at improving the effectiveness of bone marrow transplantation (BMT) include combined therapy with either genetically engineered ARSA enzyme [Martino et al 2005] or mesenchymal stem cells [Koc et al 2002, Meuleman et al 2008].
Enzyme replacement therapy (ERT) is presently considered impractical because of the difficulty of bypassing the blood-brain barrier. Clinical testing of intravenous recombinant human enzyme was discontinued in 2010 after a Phase I/II study failed to show substantial improvement [Shire 2010]. However, different forms of human ARSA enzyme are now available, and animal studies suggest that it may be a useful supplement in other therapies [Martino et al 2005, Matzner et al 2005]. Schröder et al [2010] examined N-linked glycans on recombinant ASAs produced under differing culture conditions and concluded that the enzymes used in various clinical trials may have had different uptake properties.
Gene therapy: A large number of papers have been published over the past ten years on experimentalgene therapies for arylsulfatase A. These are reviewed by Biffi & Naldini [2007], Sevin et al [2007], Biffi et al [2008b], Gieselmann [2008]. Sevin et al [2009], Faust et al [2010], Gieselmann & Krägeloh-Mann [2010], Sevin et al [2010], and Biffi et al [2011]. Piguet et al [2009] investigate intracerebral AAVrh.10 as a possible gene therapy vector for MLD. Piguet et al [2010] considered brain-directed gene therapies for MLD. Colle et al [2010] injected an adeno-associated virus vector containing human ARSA into the brains of non-human primates and found that the enzyme was expressed without adverse effects, suggesting that a similar approach could be possible in persons with MLD.
Preliminary results of a human gene replacement trial have recently been reported. The treatment appeared to be effective over an 18-24 month follow up period in the three presymptomatic affected individuals. Some concerns have been raised about the long-term safety of this approach, but the results are encouraging [Biffi et al 2013, Flight 2013, Rothe et al 2013, Verma 2013].
NCBI
Two different approaches to gene therapy are currently being researched for MLD.
Gene therapy with an autologous stem cell transplant
Italian researchers at the San Raffaele Telethon Institute tested a novel approach combining gene therapy with a stem cell transplant. Recruiting for the Phase I/II Clinical Trial formally started on March 24, 2010 after approval from the Italian Authorities. Recruiting the initial cohort of 8 patients was completed in mid-March 2013. The trial was to test the efficacy and safety of autologous hematopoietic stem cell transplantation (HSCT) after genetic modification to deliver a super-therapeutic (over-expressing) ARSA enzyme to the nervous system by the route of the blood cells. Using the patient's own stem cells with genetic correction should reduce or eliminate the complications of graft vs. host disease and provide a long term solution to proper ARSA expression in MLD patients. Bench and animal tests showed positive results. The researchers published 2-year outcomes for the first three patients in July 2013. Results were described as promising.
Intracerebral Gene therapy
A Phase I/II Clinical Trial started recruiting in Paris in late March, 2013 for an Intracerebral Gene Therapy clinical trial where special "vectors" carrying genetically modified material are directly injected into a dozen sites in the brain. The hope is that the corrected cells and the enzyme they produce will then diffuse into surrounding areas of the brain. Extensive work in the lab and some encouraging ALD studies provided the basis for this trial.
Substrate Reduction therapy
- Biomarin South from San Diego has initiated a drug discovery program for MLD. This program is based on using assays which measure sulfatide accumulation in cultured fibroblasts as a means to discover and develop small molecule drugs for MLD. As of July 2011, Zacharon has begun adapting the assays it developed for other lysosomal storage diseases so that they can be employed to discover and develop drugs for MLD.
- The Cooper Health System (New Jersey) sponsored a clinical trial underway to determine the safety and efficacy of a Vitamin K antagonist (Warfarin) in treating Metachromatic Leukodystrophy (MLD) in 2009. No results are known to have been published at March 2013.

Fig. 1 Gene marking in patients after HSC-GT.
(A) Engraftment of the transduced cells, evaluated with quantitative PCR on individual colonies from CFC assay performed on PB- and BM-derived cells and expressed as percentage (%) of LV+ colonies on total tested colonies. (B to D) VCN expressed as copies of LV/human genome measured with quantitative PCR on BM-derived CD34+ cells (B), individual subpopulations isolated from PB of patient MLD01 ©, and total PBMCs from patients MLD01, -02 and -03 (D).

Fig. 2 ARSA expression in patients after HSC-GT.
(A and B) ARSA activity measured with the p-nitrocatechol sulfate (PNC) assay on CD15+ cells (A) and CD14+ cells (B) isolated from the patients’ PB. The activity range measured in a cohort of healthy donors (HDs) (n ≥ 10 subjects) is shown. © Representative DEAE cellulose-chromatography analysis on 500 μl of cerebrospinal fluid (CSF) from a pool of four HDs, of a representative MLD patient before treatment (MLD01 pre-GT) and of the same patient 1 year after gene therapy. The peak of activity corresponds to the native form of the ARSA enzyme, as also demonstrated by its absence/very low residual activity in the patient’s pretreatment sample. (D) Specific activity (toward MUS) of the ARSA enzyme isolated from the CSF of HDs (two cohorts of four donors each) and of the treated MLD patients (circles, MLD01; blue diamond, MLD 02; and green triangle, MLD03) sampled 12 months (red circle, MLD01; MLD02 and -03 were also sampled at 12 months) and 24 months (pink circle, MLD01) after treatment.
Sciencemag
Other studies
Other studies are in progress and researches on mouse models of MLD are fundamental:
-Matzner et al [2008] evaluated parameters affecting enzyme replacement that could be an adjunct to therapy.
-Capotondo et al [2007] evaluated overexpression of ARSA.
-Hou & Potter [2009] discuss microencapsulated brain-targeted therapy for MLD.
-Lagranha et al [2008] demonstrated the ability of encapsulated BHK cells overexpressing ARSAto correct the enzyme defect in fibroblasts from persons with MLD.
“ITALIAN STUDIES”
Google
“Il virus Hiv per curare due gravi malattie genetiche: funziona l’intuizione di uno scienziato italiano. Per la prima volta nel mondo la terapia genica offre concrete speranze di cura per leucodistrofia metacromatica e sindrome di Wiskott-Aldrich. Stanno bene i sei bambini trattati a Milano”
Il virus dell’Aids può essere utilizzato per curare due gravi patologie ereditarie. Sull’intuizione “geniale” di uno scienziato italiano, avvenuta nel 1996, che negli anni aveva dato risultati promettenti in laboratorio, arriva adesso il doppio riconoscimento ufficiale da parte di una delle più importanti riviste scientifiche internazionali. E sei bambini, provenienti da tutto il mondo, dopo tre anni di trattamento stanno bene e mostrano significativi benefici. Lo annunciano due studi pubblicati oggi su Science* dai ricercatori dell’ Istituto San Raffaele Telethon per la terapia genica (Tiget) di Milano guidati da Luigi Naldini, che dimostrano che la terapia genica con vettori derivati dal virus Hiv funziona nei confronti di due gravi malattie genetiche, la leucodistrofia metacromatica e la sindrome di Wiskott-Aldrich. «A tre anni dall’inizio della sperimentazione clinica – dichiara Naldini – i risultati ottenuti sui primi sei pazienti sono davvero incoraggianti: la terapia risulta non solo sicura, ma soprattutto efficace e in grado di cambiare la storia clinica di queste gravi malattie. Dopo 15 anni di sforzi, successi in laboratorio, ma anche frustrazioni, è davvero emozionante poter dare una prima risposta concreta ai pazienti» spiega il direttore dell’ Istituto San Raffaele-Telethon di Milano.
Alla base di entrambe le malattie c’è un difetto genetico che si traduce nella carenza di una proteina fondamentale per l’organismo fin dai primi anni di vita. Nel caso della leucodistrofia metacromatica, fino ad oggi incurabile, è il sistema nervoso a essere colpito: questi bambini nascono apparentemente sani, ma a un certo punto iniziano a perdere progressivamente le capacità cognitive e motorie acquisite fino a quel momento, senza alcuna possibilità di arrestare il processo neurodegenerativo. I bambini affetti da sindrome di Wiskott-Aldrich, invece, presentano un sistema immunitario difettoso che li rende molto più vulnerabili del normale allo sviluppo di infezioni, malattie autoimmuni e tumori, oltre a un difetto nelle piastrine che è causa di frequenti emorragie. Dopo i brillanti risultati ottenuti nel corso di molti anni di studio in laboratorio, i ricercatori del Tiget hanno provato a correggere il difetto genetico che provoca queste malattie con la terapia genica. La tecnica utilizzata consiste nel prelievo delle cellule staminali ematopoietiche dal midollo osseo del paziente e nell’introduzione di una copia corretta del gene altrimenti difettoso attraverso dei vettori virali derivati da Hiv (sviluppati a partire da un primo lavoro del 1996 che porta la firma proprio di Luigi Naldini). Una volta re-infuse nell’organismo, le cellule così curate sono in grado di ripristinare la proteina mancante a livello degli organi chiave.
«Nel caso della leucodistrofia metacromatica, invece – dice Alessandra Biffi, a capo dell’altro studio – il meccanismo terapeutico è più sofisticato: le cellule ematopoietiche corrette raggiungono il cervello attraverso il sangue e lì rilasciano la proteina corretta che viene “raccolta” dalle cellule nervose circostanti. La carta vincente è stata quella di rendere le cellule ingegnerizzate in grado di produrre una quantità di proteina molto superiore al normale, in modo contrastare efficacemente il processo neurodegenerativo». Aggiunge Eugenio Montini, che ha coordinato le analisi molecolari sulle cellule dei pazienti: «Non avevamo mai visto finora una ingegnerizzazione delle cellule staminali attraverso la terapia genica così efficace e sicura. Questi risultati aprono la strada a nuove terapie anche per altre malattie genetiche e più diffuse».
Milano, 11 luglio 2013
Lentiviral hematopoietic stem cell gene therapy benefits metachromatic leukodystrophy
Discussion
Our gene therapy protocol allows stable engraftment of transduced HSCs at high levels and without evidence of vector-induced genotoxicity. The reconstitution of ARSA activity in the cerebrospinal fluid and the arrested progression of neurodegenerative disease in the three treated patients demonstrate that the transplanted cells, or their progeny, can seed the nervous system and deliver therapeutic levels of active enzyme. Although our data are promising, long-term follow-up of the patients is needed in order to establish the full therapeutic potential of this gene therapy strategy for MLD. In addition, our data position LV gene transfer as a feasible means to engineer human hematopoiesis to its near entirety—an approach that could be exploited for treatment of other diseases.
Science
 Fig. 3 Clinical follow up of MLD patients after HSC-GT.
Fig. 3 Clinical follow up of MLD patients after HSC-GT.
(A and B) GMFM score (A) and NCV index (B) of the three treated patients and of a historical cohort of LI-MLD patients (gray circles). The dotted lines indicate for each treated patient (inset, color code) the expected time of disease onset, according to the disease onset observed in their affected matched siblings; n.r., normal range of the NCV index. © Axial T2 weighted fast spin-echo MR images (top) and FLAIR MR images (bottom) obtained from patient MLD01 at baseline (before GT) and at +2 years after treatment, and corresponding (equivalent) images of an age-matched untreated patient with LI-MLD (in parenthesis, the chronological age at imaging acquisition in months). In MLD01 images, a small area of hyperintensity is present within the splenium of the corpus callosum, stable in extension and appearance as compared with that in the +12 months follow-up; subtle signal inhomogeneities are present within posterior periventricular and posterior centrum semiovale white matter, in the absence of focal lesions; these inhomogeneities, present since baseline as just barely T2 and FLAIR hyperintensity signal, are (now) more evident because of the normal signal of the surrounding myelin; subarachnoid and ventricular spaces are within normal limits, even if a little wider when compared with that of the baseline. Basal ganglia and thalami remain of normal appearance. In UT LI-MLD images, extensive, diffuse symmetric hyperintensities with typical “tigroid pattern” are seen within periventricular white matter, centrum semiovale, corpus callosum, external and internal capsules, and cerebellar deep white matter. A severe diffuse brain atrophy involving basal ganglia and thalamy, which show T2 hypointense signal, is also present.
Sciencemag
VIDEO INTERVIEWS WITH THE PRINCIPAL INVESTIGATORS;
mldfoundation
Fonti
Orphanet
Wikipedia
Google
National Center for Biotechnology Information
NCBI pubmed
NCBI pubmed
NCBI pubmed
American Journal of Neuroradiology
AJNR
AJNR
Nature