Giulia Mezzacappa
Ilenia Ferraro
ROHHAD SYNDROME
Rapid-onset obesity with hypothalamic dysfunction, hypoventilation, and autonomic dysregulation (ROHHAD) is a rare disorder of respiratory control and autonomic nervous system (ANS) regulation, with endocrine system abnormalities. It mimics both common obesity and genetic obesity syndromes along with several endocrine disorders during early childhood.
ROHHAD presents after 1.5 years of age, in otherwise healthy children. The rapid-onset weight gain (often 30 pounds in 6-12 months) is typically the herald of the disease and the harbinger of the later features of the ROHHAD phenotype. The acronym ROHHAD describes the typical sequence of symptoms experienced by most children with ROHHAD, in the order of their appearance. The clinical features of ROHHAD seem to “unfold” with advancing age in each child.

EPIDEMIOLOGY
ROHHAD was first described in 1965 (albeit under a different name) and since that time at least 100 children have been reported in the literature. Because of the high prevalence of cardiorespiratory arrest, early recognition and treatment of the symptoms associated with ROHHAD are essential and may be life-saving.
ROHHAD is a relatively "new" disorder without many cases identified thus far, it is not yet clear if any certain population is at greater risk for developing ROHHAD. Because of the explosion of exogenous obesity worldwide, a very high level of vigilance in consideration of ROHHAD is essential.
SYMPTOMS
- 1) RO (Rapid-onset Obesity)*
Children with ROHHAD are seemingly normal before the rapid-onset weight gain. Between 1.5 and 7 years of age, these children begin to manifest abnormalities that will evolve into the features of ROHHAD. Most commonly, the first sign is dramatic (often 20-30 pounds) and rapid (over 6 to 12 months) weight gain with associated abnormal increase in hunger (hyperphagia). This rapid-onset obesity could be considered a sign of hypothalamic dysfunction.
- 2) H (Hypoventilation)*
After the rapid weight gain, children with ROHHAD will begin to show breathing abnormalities. Some children will have obstructive sleep apnea; they may have snoring and they may have pauses in breathing related to the air flow blockage (obstructive apnea). All children with ROHHAD develop alveolar hypoventilation with very shallow breathing during sleep (nap and night). In more severely affected patients with ROHHAD, the hypoventilation is apparent awake and asleep. In patients with ROHHAD, a lack of normal responsivity to low oxygen and elevated carbon dioxide occurs during wakefulness as well as sleep, even when awake breathing is adequate. This hypoventilation/control of breathing deficit is the most life-threatening feature of ROHHAD, yet it is often unnoticed until after a dramatic event such as a cardiorespiratory arrest.
- 3) H (Hypotalamic Disfunction)*
Other hypothalamic abnormalities may not be detected at the time of the rapid weight gain, but will be identified later. These other hypothalamic/endocrine abnormalities may include inability to maintain normal water balance in the body (leading to abnormally high or low sodium levels), high prolactin levels, low thyroid hormone, early or late puberty, and low cortisol among other abnormalities. . Patients with ROHHAD can become dehydrated due to lack of thirst with normal or partial antidiuretic hormone Secondary also narcolepsy has been described as a neurologic disorders due to hypothalamic dysfunction function. Children with ROHHAD can have variable timing and number of these symptom, but all will have some abnormalities.
- 4) AD (Autonomic Disfunction)*
These include eye abnormalities such as altered pupil response to light, “lazy eye” (strabismus), intestinal abnormalities such as altered motility which causes chronic constipation or diarrhea, temperature dysregulation with episodes of very high body temperature (hyperthermia) and altered sweating or more typically very low body temperatures (hypothermia), decreased sensation of pain, low heart rhythm that may be so slow that a cardiac pacemaker is required, icy cold hands and feet, and many other symptoms reflecting dysregulation of automatic functions.
ROHHAD-NET
Some individuals (40%) with ROHHAD will develop tumors of neural crest origin like ganglioneuromas or ganglioneuroblastomas. They seem to be associated with polyalanine repeat expansion PHOX2B mutations (PARMs) , but more commonly with the non-PARMs (NPARMs) (~40% of cases have a neuroblastoma).
Other features in a subset of individuals with ROHHAD include behavioral, mood, and developmental disorders such that the Intelligence Quotient (IQ) score may be reduced. However, a remarkably high percentage of children with ROHHAD have normal or high IQs, suggesting that IQ in children with ROHHAD likely has many related factors. Preliminarily, those children with the behavioral and mood disorders have more typically had suboptimal ventilatory support. Some children with ROHHAD may develop seizures, though this feature may be related to episodes of low oxygen (hypoxemia) levels due to inadequate ventilator support.
PATHOGENESIS
The diagnosis of ROHHAD is currently based on clinical criteria and though investigation of genetic mutations is underway, no specific cause for ROHHAD has been found to date.
Since the first published description in 1965, ROHHAD has often been associated and confused with Congenital Central Hypoventilation Syndrome CCHS. Howevr, the clinical distinction of ROHHAD from CCHS coupled with the discovery of mutations in the paired-like homeobox 2B (PHOX2B) gene as disease defining in CCHS. The subsequent lack of these mutations in ROHHAD subjects established the distinction between these two clinical entities.
Genetic hypotesis. The constellation and consistency of symptoms and presentation of ROHHAD, combined with reported familial recurrence, suggest a monogenic cause. However, previous candidate gene studies have failed to identify a disease-associated genetic variation and are limited by small sample size. Investigations have focused on genes involved in development and function of the hypothalamic, autonomic, and/or neuroendocrine systems due to deficiencies of these systems in ROHHAD.
The 5-hydroxytryptamine (serotonin) receptor 1A (HTR1A) shows hypothalamic expression and has a known role in appetite control and energy regulation. Furthermore, this gene is densely distributed in medullary regions important for cardiorespiratory regulation and has a key role in the autonomic response to homeostatic stress.
Like HTR1A, the orthopedia (OTP) gene shows hypothalamic expression, with an important role in hypothalamic cell specification in the developing hypothalamus. The paraventricular nucleus complex of the hypothalamus, important in homeostatic control functions, has also been shown to express OTP.
Pituitary adenylate cyclase activating polypeptide (PACAP) is expressed in brain regions important to respiratory, cardiovascular, visceral, and thermoregulatory control including the paraventricular nucleus complex of the hypothalamus. PACAP has also been shown to be essential for normal lipid and carbohydrate metabolism as well as maintenance of normal energy homeostasis. PACAP also plays an important role in respiratory chemosensitivity and preventing neonatal hypoventilation at reduced body temperatures.
The HTR1A, OTP, and PACAP genes were targeted as candidate genes in the etiology of ROHHAD based on their roles in the embryologic development of the hypothalamic, autonomic and/or neuroendocrine systems. In addition, these genes have known roles in homeostasis, respiratory control, metabolism, and thermal regulation, all processes which appear to be dysfunctiona in ROHHAD.
Variations within coding regions of the HTR1A, OTP, and/or PACAP genes would be associated with ROHHAD. Thirteen variations were identified within this cohort of 25 ROHHAD cases and 25 matched controls. However, none of the variants identified in the studies was significantly associated with ROHHD.
Identification of monozygotic twins discordant for ROHHAD does not preclude genetic variation as a causative factor in ROHHAD, and might involve modifier genes in the variable phenotype, it also indicates that stronger consideration must be given to alternative etiologies.
Variation in the epigenomes of identical twins has been shown to accumulate quickly and throughout their lifetime, accounting for a great deal of discordance in phenotypes of twins. Variation in the epigenome has previously been found to plays a vital role in development of pediatric diseases of respiratory and autonomic function such as Prader-Willi and Rett Syndrome. The possibility that epigenetic variation plays a role in the development of ROHHAD will be analyzed in future studies.
Autoimmune and paraneoplastic mechanisms. Particular consideration has been given to the possibility that ROHHAD may be a paraneoplastic condition based on the finding that neural crest tumors occur in 30–50% of reported cases.
Paz-Priel et al. described a single case with some symptoms suggestive of ROHHAD in which highdose cyclophosphamide was given with broad clinical improvement. As this case did not present with the hypoventilation typical of ROHHAD, it is not clear how relevant this result can be to the broader ROHHAD population. The finding does support the need for additional analysis of the hypothesis that ROHHAD may be a paraneoplastic condition, at least in a subset of cases. However, tumors have not been identified in more than 50% of reported ROHHAD cases. In addition, tumor removal in the subset of ROHHAD cases with tumors has failed to alter the other features of the phenotype, and immunosuppressive strategies have produced mixed results. Furthermore, the autoantibodies in serum and cerebrospinal fluid that characterize paraneoplastic syndromes have not yet been identified in the few cases of ROHHAD studied.
It is also possible that ROHHAD represents an autoimmune phenomenon precipitated by specific environmental exposures in an individual with a susceptible genetic background, as has been postulated for narcolepsy. This alternative could explain the variable timing of onset in the disease
Hypocretin 1. Karlien Dhondt et al for the first time describe a patient with ROHHAD syndrome with a secondary form of narcolepsy/cataplexy associated with a hypocretin deficiency. This extension of the ROHHAD phenotype may have therapeutic opportunities, although the possible role of stimulants or anticataplectic therapy remains to be established. Further research should focus on the presence and clinical presentation of narcolepsy/cataplexy features in children with ROHHAD.
Several features are in common between the two syndromes but the absence of other hypothalamic and hormonal dysfunctions in narcolepsy remains an important differential sign.
The hypocretins play an important role in central cardiovascular regulation, enhancing sympathetic outflow and plasma catecholamine release and a reduced sympathetic tone is observed in narcoleptic patients. In ROHHAD, autonomic manifestations were present in all studied patients and appear quite early, at disease onset. In part, these symptoms may relate to hypocretin dysfunction.Metabolic changes also appear in the early stage of both narcolepsy and ROHHAD.
In narcolepticpatients some authors suggested that overweight could be a clinical manifestation of the underlying autonomic dysfunction.Shen showed that highdose hypocretin administrated to rats results in lipolysis, whereas lack of hypocretin signaling inhibits lipolysis and leads to fat storage. In both narcolepsy and ROHHAD, lipogenesis may thus be surpassing lipolysis.
An autoimmune-mediated process has been speculated as a possible cause of ROHHAD.
In narcolepsy, an autoimmune cause is suspected too, fitting with the selective loss of hypocretin-producing neurons and the strong HLA association: 95% of narcolepsy patients carry the DQB1*0602 allele. This particular allele was not found by Pontual et al in patients with ROHHAD.However, the hypothesis of autoimmunemediated destruction of hypocretin neurons in ROHHAD remains tantalizing, as hypocretin peptides are expressed in ganglioneuroblastomas, providing a potential immune target.
DIAGNOSIS
The criteria for diagnosis of ROHHAD include the following:
1) Rapid-onset obesity and alveolar hypoventilation during sleep starting after the age of 1.5 years,
2) Evidence of hypothalamic dysfunction, as defined by at least 1 of the following findings: rapid-onset obesity, hyperprolactinemia, central hypothyroidism, disordered water balance, failed growth hormone stimulation test, corticotrophin deficiency, or altered onset of puberty (delayed or precocious)
3) Absence of a CCHS-related PHOX2B mutation (to genetically distinguish ROHHAD from CCHS). At present there is no genetic testing available to diagnose ROHHAD, so the diagnosis is based on the clinical presentation and clinical course which should include cooperative consultation by experts.
CLINICAL TESTING
Initial evaluation can include overnight polysomnography to evaluate for any signs of obstructive sleep apnea during sleep.
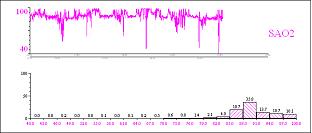
Diurnal hypoventilation can be diagnosed by monitoring SpO2 and CO2 levels during the day. Detection of SpO2 < 95% and CO2 > 50 mmHg allows the diagnosis of vigil hypoventilation.
Sequential comprehensive evaluation is recommended, ideally at a Center with expertise in respiratory and autonomic medicine--including expertise specifically with ROHHAD. This evaluation includes> detailed respiratory physiologic evaluation while awake and while asleep, comprehensive cardiac evaluation including 72-hour Holter, cycle ergometry (exercise test), and echocardiogram, neurocognitive testing for tracking intellectual function as a marker for neurologic stability vs. decline, endocrine evaluation for development of new symptoms of hypothalamic dysfunction, and age-appropriate evaluation of ANSD. Important is also imaging of chest and abdomen to screen for evidence of neural crest tumors
A complete evaluation of endocrine function should be performed with particular attention to water balance regulation, obesity, and other signs of pituitary dysfunction. If hypernatremic dehydration is found, formal testing of antidiuretic hormone secretion should be done before assuming the patient has diabetes insipidus. Obesity can alter growth hormone secretion and levels of insulin-like growth factor-1 (IGF-1) which should be taken into account when assessing growth hormone function. Complications of obesity including fatty liver, elevated lipids, or diabetes mellitus should be considered.
TREATMENT
The treatment of ROHHAD at present is based on the clinical features and their relative severity.

The obesity is exceedingly difficult to control with diet and exercise. More effective is special emphasis to avoid further weight gain as the child grows vertically (with or without growth hormone supplement to treat growth hormone deficiency), but intervention requires consultation with a nutritionist and endocrinologist. Since patients with ROHHAD do not increase their breathing adequately during physical exertion, it is important to recommend only modest exertion until safe parameters have been established by a physician based on end tidal carbon dioxide and pulse oximetry monitoring during exercise.
The hypothalamic dysfunction seen in these patients must be evaluated and treated by a pediatric endocrinologist. Teatments may include hormone replacement, a strict fluid intake regimen, and other measures designed to make up for a dysfunctional hypothalamus. Growth hormone administration on the basis of failed stimulation testing has not been shown to improve body composition and use of dopamine agonist to normalize prolactin levels has not been shown to modify the clinical course.
One of the main challenges in ROHHAD is the control of breathing deficit that is typically unapparent at the time of the rapid-onset weight gain, but seems to worsen with advancing age in many children. Some children may initially need artificial ventilation during sleep only, then progress to need for continuous support (awake and asleep). From the beginning, the key goal is optimization of oxygenation and ventilation. Many patients with ROHHAD can be managed with mask ventilation and bi-level positive airway pressure at night only (but provided with an actual mechanical ventilator); those children requiring 24 hour/day mechanical ventilation will need a tracheostomy through which the patient is then mechanically ventilated. These children require a mechanical ventilator at home as well as experienced registered nursing care ideally 24 hours/day.
One report describes transient response to conventional immunosupression combined with rituximab and a sustained response to high-dose cyclophosphamide. On two separate occasions, the patient experienced symptomatic improvement shortly after initiation of rituximab therapy, but her symptoms recurred with waning of the immunosuppressive effect. Alonger remission was achieved after high-dose cyclophosphamide induced immunoablation. The pattern that they describe fulfills the temporality, consistency, and doseresponse criteria of causality. Moreover, an autoimmune process is biologically plausible, given that neuroblastoma is associated with autoimmune-mediated paraneoplastic syndromes.
Thus, patient’s course indicates, albeit indirectly, an autoimmune mechanism in ROHHAD. However, ROHHAD syndrome may include etiologically diverse subgroups of patients, and autoimmunity may not be universally relevant.
Spontaneous improvement cannot be excluded, it is less likely in this patient, who experienced continuous deterioration except for the three occasions when initiation of immunosuppression was associated with rapid amelioration of ROHHAD manifestations.
INVESTIGATIONAL THERAPIES
An International ROHHAD Registry has become clinically available for all patients with the clinical diagnosis of ROHHAD. The purpose of the Registry is to collect data about the clinical development of ROHHAD with advancing age as the disease process unfolds . The Registry is achieved through a secure questionnaire via REDCap (Research Electronic Data Capture). The aim is to consent and enroll ALL patients worldwide with ROHHAD in order to have a central repository to advance understanding of this rare disease and improve early diagnosis, offer anticipatory management, and decrease disease burden.
BIBLIOGRAPHY
1. raredisease.org
2. Rapid-Onset Obesity With Hypothalamic Dysfunction, Hypoventilation, and Autonomic Dysregulation: Analysis of Hypothalamic and Autonomic Candidate Genes. 2011
3. Monozygotic Twins Discordant for ROHHAD Phenotype. Pediatrics 2011;128;e711; originally published online August 1, 2011
4. Hypocretin-1 Deficiency in a Girl With ROHHAD Syndrome. Pediatrics 2013;132;e788; originally published online August 12, 2013