Martina Cortese
Cardiomyopathies are defined by structural and functional abnormalities of the ventricular myocardium that are unexplained by flowlimiting coronary artery disease or abnormal loading conditions.
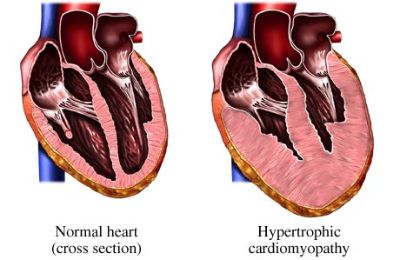
Hypertrophic cardiomyopathy (HCM) is defined by the presence of increased left ventricular (LV) wall thickness that is not solely explained by abnormal loading conditions.
In up to 60% of adolescents and adults with HCM, the disease is an autosomal dominant trait caused by mutations in cardiac sarcomere protein genes. Five to ten percent of adult cases are caused by other genetic disorders including inherited metabolic and neuromuscular diseases (Anderson Fabry disease, Friedrich’s ataxia), chromosome abnormalities and genetic syndromes (Noonan, LEOPARD). Some patients have non-genetic disorders that mimic genetic forms of the disease, for example, senile (TTR) and (AL) amyloidosis.
Mutations in the genes encoding beta-myosin heavy chain (MYH7) and myosin-binding protein C (MYBPC3) account for the majority of cases; less commonly affected genes include cardiac troponin I and T (TNNI3, TNNT2), tropomyosin alpha-1 chain (TPM1) and myosin light chain 3 (MYL3). In general, patients with a sarcomere protein mutation present earlier and report a higher prevalence of family history of HCM and sudden cardiac death (SCD) than those without a mutation. They also tend to have more severe hypertrophy, microvascular dysfunction and myocardial fibrosis.(1)
Galectin – 3
Galectin-3 (Mac-2, CBP-35, eBP, RL-29, HL-29, L-34, LBP) is a 29– 35 kDa chimaera-type galectin which is unique in that it is the only member of the galectin family with an extended N-terminal domain constituted of tandem repeats of short amino acid segments (a total of 110–130 amino acids) linked to a single C-terminal carbohydrate-recognition domain of about 130 amino acids. Whereas the C-terminal domain is responsible for lectin activity, the presence of the N-terminal domain is necessary for the full biological activity of galectin-3.

Galectin-3 interacts with numerous ligands: carbohydrates, such as N-acetyllactosamine (LacNac) and unglycosylated molecules, such as cell surface receptors (macrophage CD11b/CD18) and extracellular receptors (collagen IV). Binding to carbohydrates usually involves the C-terminal domain, whereas binding to unglycosylated molecules involves the N-terminal domain.
When secreted into the extracellular space (via a non-classical secretory pathway that circumvents the endoplasmic reticulum and Golgi complex), galectin-3 can interact with cell surface receptors and glycoproteins to initiate transmembrane signaling pathways for different cellular functions.
Galectin-3 is necessary for normal phagocytic activity and it has been localized in the phagocytic cups and phagosomes of macrophages.
Galectin 3 in hypertrofic cardiomiopathy and heart failure
In normal rat, murine and human hearts, the expression of galectin-3 is low. However, as the cardiac failure progresses, galectin-3 becomes rapidly and significantly up-regulated.
In hypertrophied hearts and during active myocarditis, significant infiltrations of activated macrophages were observed and galectin-3 was found to be co-localized with macrophages. These observations suggest a galectin-3- dependent stimulatory effect for macrophage migration.
Galectin-3 binding sites were localized around the nucleus of proliferating fibroblasts, whereas resting cells only had minimal binding sites in the cytoplasm. It was suggested that galectin-3 induced cardiac fibroblast production via the activation of cyclin D1.
At the site of injury, galectin-3 is secreted into the extracellular space, which contributes to the fibrotic process by activating resting fibroblasts into matrix producing fibroblasts. Fibroblast activation is characterized by the increased expression of the cytoskeletal protein a-smooth muscle actin (a-SMA, an intracellular fibrosis marker) and the extracellular type 1 collagen a-1 chain (COL1A1, an extracellular fibrosis marker). Both a-SMA and COL1A1 have been shown to be up-regulated in fibrotic tissues via the galectin-3-mediated activation of cyclin-dependent kinase inhibitor 1A, inhibitin beta A and fibronectin 1, and extracellular signal-regulated kinase and phosphatidylinositol 3-kinase, respectively.
Galectin-3 not only affects the synthesis of new matrix components such as type I collagen, it also influences the degradation of extracellular matrix components through a set of tissue inhibitor metalloproteinases (TIMPs) and matrix metalloproteinases (MMPs).

Role of galectin-3 in cardiac remodeling, fibrosis and inflammation
Experimental data and observations underscore the potentially important role that galectin-3 may play in cardiac remodelling, and interference with galectin-3 levels seems to effectively reverse cardiac remodelling. On the other hand, there is no definite proof that galectin-3 is causally involved in the pathophysiology of cardiac remodelling.
Fibrosis and scar formation are part of the maladaptive cardiac response to injury. Fibroblasts and myofibroblasts as well as macrophages have been identified as key cells in the initiation and progression
of tissue scarring. The up-regulation of galectin-3 has been demonstrated in different human fibrotic conditions, such as in liver cirrhosis, idiopathic lung fibrosis, and chronic pancreatitis.
Galectin-3 mRNA expression was significantly correlated with the extent of fibrosis.
Inflammation is a prerequisite for tissue healing and scar formation.
However, when inflammation becomes sustained it can lead to the formation of extensive scar tissue, leading ultimately to complete organ failure. It has been suggested that macrophages are the key
cell type in the development of fibrosis.
Macrophage secretion and galectin-3 expression are major mechanisms in myofibroblast accumulation and activation, and subsequent renal and cardiac fibrosis. (2)
HF therapy aimed at inflammatory responses may need to be targeted at the early stages of HF and probably needs to antagonize multiple inflammatory mediators, including galectin-3. (3)
Galectin 3 in clinical practice
Left ventricular (LV) remodeling in heart failure is related to deterioration of myocardial performance and to worse outcome. LV fibrosis, assessed by late gadolinium enhancement (LGE) at cardiac magnetic resonance (CMR) imaging, is a marker of LV remodeling, and holds prognostic value in nonischemic dilated cardiomyopathy (DCM). Galectin-3 has been recently shown to participate in tissue fibrogenesis and to be
useful as prognosticator in heart failure patients.
Galectin-3 levels, even lower than previously reported prognostic cut-off, predict the presence of myocardial replacement fibrosis, assessed by LGE technique, in patients with nonischemic DCM. This results support the hypothesis that galectin-3 is involved in cardiac fibrosis and remodeling and that may help to select high risk subsets among heart failure patients. (4)
Similarly to DCM, it would be important to assess a correlation between Gal-3 levels and LGE in CMI because this CMR parameter holds prognostic value. Some patients affected by the disease cannot undergo CMR because of contraindications, such as pacemakers, claustrophobia. Especially in these cases it would be extremely useful to find new parameters, such as Gal 3 blood testing, to replace the radiologic exam.
Galectin 3 levels measured in blood are influenced by patient’s age, sex, GFR and some drugs. Because of its role in mediating inflammation related fibrogenesis, it is quite hard to discriminate among Gal-3 raises related to cardiac fibrogenesis in CMI and development of fibrosis in other organs, such as lung in COPD and cirrhotic liver. Raised level in Gal 3 have been found in patients with worse prognosis, but this could be explained by a worse general clinical condition independent from cardiac fibrosis, such as comorbilities.
Gal 3 is a promising marker involved in CMI pathogenesis but further studies are needed to characterize Gal 3 pathway, establish his prognostic role and correlation with other parameters and exams used in CMI diagnosis and follow up.
(1) (European Heart Journal
)
(2) (Galectin-3: a novel mediator of heart failure development and progression.
)
(3) (Galectin-3 marks activated macrophages in failure-prone hypertrophied hearts and contributes to cardiac dysfunction.)
(4) (GALECTIN-3 AND MYOCARDIAL FIBROSIS AT CARDIAC MAGNETIC RESONANCE IN NONISCHEMIC DILATED CARDIOMYOPATHY)