DEFINITION
Kidney disorder with autosomal dominant inheritance and characterized by multiple cysts in both kidneys with progressive deterioration of renal function. The disease database
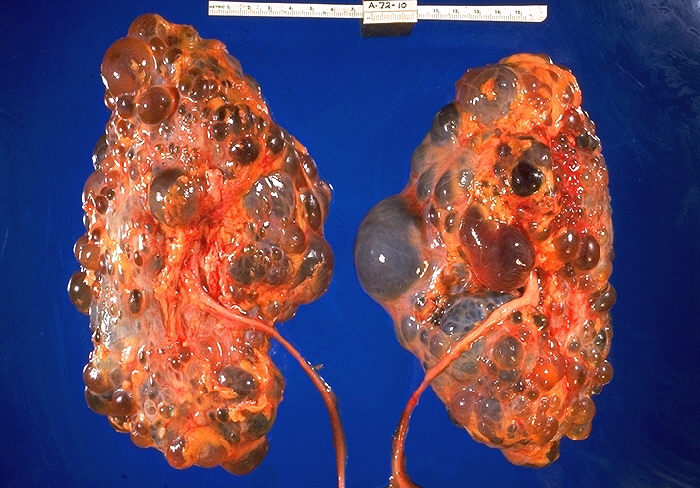
EPIDEMIOLOGY
Polycystic kidney disease is the most common life-threatening genetic disease, affecting approximately 7 million people worldwide. Autosomal dominant polycystic kidney disease (ADPKD) affects about 1 in 1000 (1:400–1:1200) persons in Caucasian populations and may be 2.5–15 times more frequent than other common hereditary disorders such as cystic fibrosis and sickle-cell disease. It occurs equally in men and women.
SYMPTOMS
It is typically a late onset disorder with renal failure developing in the fourth or fifth decade in the case of autosomal dominant polycystic disease type 1 (PKD1), or a decade later in the case of autosomal dominant polycystic disease type 2 (PKD2), in approximately 50% of patients. Considerable clinical variability in the age of onset of renal failure is observed within and between families.
Most common symptoms are not specific and shared with other diseases .
DIAGNOSIS
ADPKD is typically diagnosed in adults by the detection of bilaterally enlarged polycystic kidneys using trans-abdominal ultrasound scanning.Computed tomography (CT) and magnetic resonance imaging (MRI) are also used where additional information on renal structure and function are required or where ultrasound resolution is poor such as in the obese individual.
Hypertension may be demonstrated in 60 per cent of patient.
Urinary findings: albuminuria, haematuria, low urinary density, elevated BUN
PATHOGENESIS
Two proteins, polycystin 1 (the PKD1 gene product) and polycystin 2 are central to this condition with PKD1 mutations accounting for 85% of all ADPKD cases and PKD2 responsible for the majority of the rest.
PC-1 and -2 are joined via a domain in the carboxy-tail of PC-1, and appear to act in concert.
Similar symptoms dependd on different genes, probably via some common pathways.
Acquired PKD can occur in chronic renal failure of any cause and is common in the dialysis population. Other polycystic kidney diseases include multicystic dysplastic kidney, autosomal recessive polycystic kidney disease, tuberous sclerosis, von Hippel– Lindau disease, nephronophthisis type 2, oro-facial digital syndrome type I and disease due to mutations in the HNF1b gene.
mTOR pathway
mTOR in VHL pathway
interaction between mTOR and TSC
In addition to renal cysts, cysts occur in
- the liver (70%)
- and pancreas (5–10%).
1 Cysts have also been reported in other organs such as the spleen, thyroid, arachnoid, seminal vesicles and prostate. Noncystic manifestations of
ADPKD are also common. These include hypertension (70–80%), cardiac valve anomalies (25%) and intracranial vascular abnormalities including vascular aneurysms (8%).
PATIENT RISK FACTORS
Genetic
In the PKD1 gene, the location of the mutation appears to influence the disease severity with mutations in the 5’ portion of the gene (0–7812 nt) associated with a lower mean age of ESRF than those 3’ of 7812 nt.
Hormonal
There is no gender effect on CKD progression after the GFR begin to fall.
Testosterone influences the progression of renal cystic change in male and female rats with ADPKD.
TISSUE SPECIFIC RISK FACTORS
Anatomical
Some anatomical aberration like pyelo-uretheral junction disease or cisto-uretheral reflux promote infections and thus ckd progression.
Vascular
Hypertension is an important progression factor.
Some evidences suggest that the circulating RAS is not of primary importance. However, this does not exclude a role for the intra-renal RAS, which may be activated and disregulated in ADPKD, and could thereby contribute to the development of hypertension.
Physiopathological
Some evidences suggest that interstitial inflammation may be important in the development of ESRD in ADPKD: monocyte chemoattractant protein-1 (MCP-1) and osteopontin mRNAs and proteins are elevated in kidneys of animal pkd model.
UTI, macroscopic and microscopic haematuria and proteinuria are associated with progression.
Gender, gender of parent who transmitted PKD1, family history of essential hypertension, multiparity, and use of the OCP were not identified as risk factors for renal events in PKD.
COMPLICATIONS
About 50 percent of patients with PKD will have kidney failure by age 60, and about 60 percent will have kidney failure by age 70.
- Certain people have an increased risk of kidney failure including:
- men
- patients with high blood pressure
- patients with protein or blood in their urine
- women with high blood pressure who have had more than three pregnancies.
- Other clinical predictors of more severe renal disease and progression to renal failure include:
- early age of diagnosis
- early age of diagnosis of hypertension
- hepatic cysts in women
- high parity
- urinary tract infection
- macroscopic haematuria
- renal size expressed as renal volume.
The main complications associated with renal cysts include renal failure, cyst infection and haemorrhage, renal stones and pain.
FOLLOW-UP
ADPKD is a very slowly evolving condition, and GFR is well maintained until relatively late in the course of the disease. It is well recognised that deterioration of renal excretory function in ADPKD follows a unique time course in that glomerular filtration rate remains constant for many years and only declines relatively late in the course of the disease, when mechanical compression of normal renal tissue by cysts occurs. Thus, use of GFR alone as a marker of disease progression over relatively short time periods is problematic, and ideally would require following patients for many years, which is generally not feasible. A better indicator of disease progression than GFR would be successive evaluation of cyst size. Kidney enlargement is continuous and quantifiable by MRI, and higher rates of enlargement are associated with more rapid decreases in renal function.
THERAPY
Hypertensive control: ACEIs and ARBs, not diuretics(concentrating defect in pkd). In ADPKD patients ACE inhibition appeared to result in slower deterioration in renal function.
Use of statins: cell proliferation is modulated via ras oncogenes, and ras production is increased in PKD. Farnesyl-pyrophosphate, an intermediate in the conversion of acetyl Co-A to cholesterol, is required for the activation of ras-GTP-binding proteins, which are important in a number of cell functions, including cell proliferation. Statins reduce farnesyl production, and thus potentially ameliorate the accelerated cellular proliferation in PKD.
Sirolimus: inhibits the protein kinase mTOR, which appears to be a major target of cell growth and proliferation.
V2 receptor inhibition: vasopressin is the main stimulator of adenylyl cyclase, and thus of cAMP production in these cells.
C-myc antisense oligonucleotide, EGFR tyrosine kinase inhibitors and Octreotide are in study.
When ESRF is established, the therapeutical options are dialysis and transplant.
Written by Marcello Capella and Federica D'Amico.
dott Vilma Mantovani Bologna
per quanto riguarda l'analisi genetica del rene policistico, mi
risulta che i geni PKD1 e PKD2 vengano analizzati nella genetica
molecolare del Policlinico sant'Orsola-Malpighi di Bologna
(responsabile del laboratorio dottoressa Vilma Mantovani, direttore
della struttura Marco Seri). Utilizzano il sequenziamento di nuova
generazione (NGS) con tempi teoricamente più rapidi di altri centri
(san Raffaele di Milano). da Cecilia Bracco