DEFINITION
Fibrinogen, known as Coagulation Factor I, is an esameric glycoprotein of 340 KDa composed of three pairs of non-identical polypeptide chains called Aα, Bβ and γ.
It’s synthesized by parenchimal hepatic cells and it’s found as a soluble protein in human plasma, in variable concentration of 160 to 400 mg/dl and as deposit in platelets’ alpha-granules. Genes FGA, FGB and FGG encoding for chain Aα, Bβ and γ, respectively, are located, in single copy and adjacent positions, in chromosome 4 (q31.3-q32.1), are ca. 6 (FGA), 8 (FGB) and 10.5 (FGG) Kb large and form a cluster of ca. 50Kb.
THE GENES
Genes FGA and FGG are oriented in the same direction and encoded in the opposite direction of FGB. Gene FGA has 6 exons and 5 intronic regions and at the sixth exon sustains an alternative splicing that encodes for 2 proteins: the first one is the chain Aα precursor of 644 amino acids, the second is an 866 amino acids polypeptide called isoform alpha-E (extended). Gene FGB has 8 exons and 7 intronic regions and encodes for a 491 amino acids polypeptide. Finally, FGG has 10 exons and 9 intronic regions and encodes for 2 isoforms by alternative splicing of exon 10, a predominant one of 437 amino acids and an alternative one of 453 amino acids.
CHEMICAL STRUCTURE AND IMAGES

Protein Amino acids Percentage


SYNTHESIS AND TURNOVER

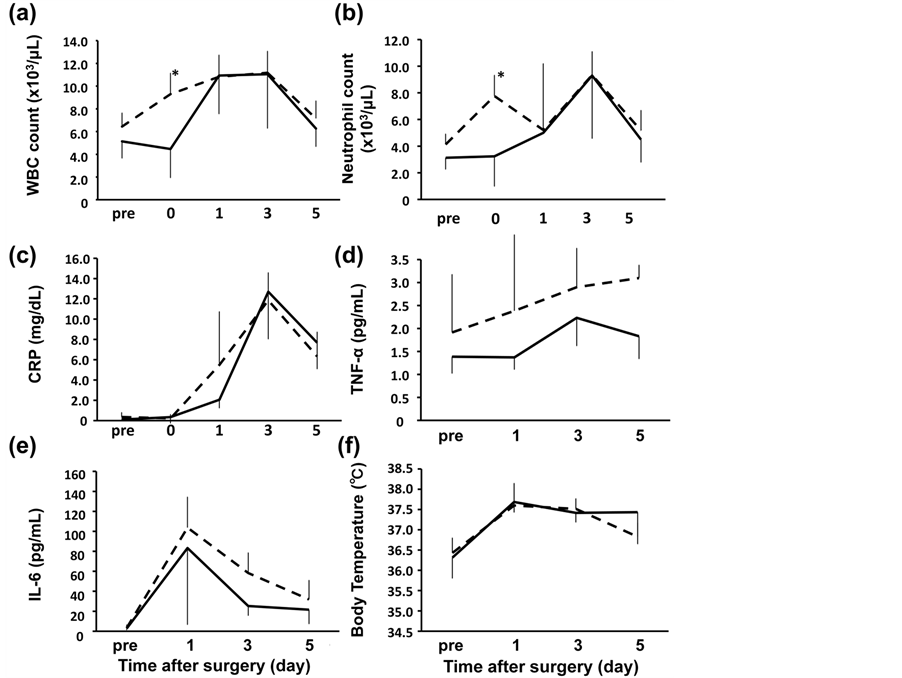


Early time course of the acute phase protein response in man. 1983
Acute Phase Proteins
CRP and fibrinogen mRNA synthesis is upregulated by IL-6 through STAT3 activation
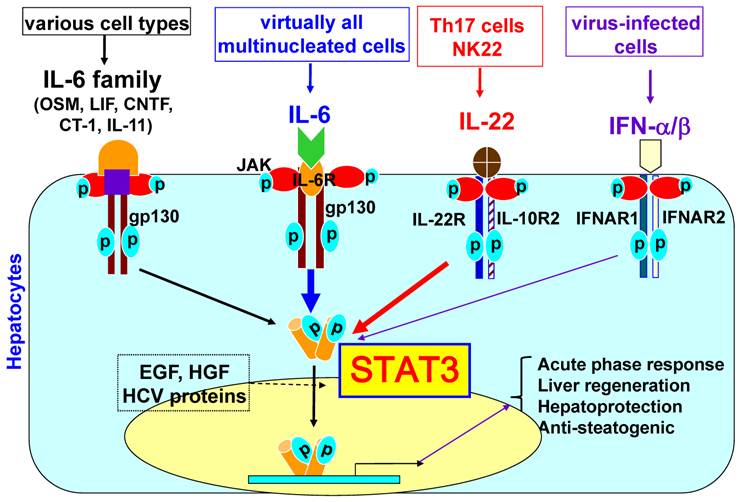
All molecules involved in STAT3 signaling have the same Trp %

protein synthesis
STAT3 INHIBITORS

post-translational modifications
degradation
The 3 primitive chains have a signal peptide of 19, 30 and 26 amino acids, respectively, that enables the translocation to rough endoplasmic reticulum where a signal peptidase removes it allowing the forming of 625, 461 and 411 amino acid mature chains.
The six chains assembly to form the esameric protein via an elaborate process that provides the initial dimeric association of Aα-γ and Bβ-γ polypeptides; subsequently there is the association of the trimeric intermediate protein Aα-Bβ-γ and finally the forming of the esameric protein (Aα-Bβ-γ) 2 . The forming of 12 intrachain disulfuric bridges and 17 interchain disulfuric bridges stabilizes the tertiary structure.
After the assembling, fibrinogen has a linear array of 3 nodules held together by a very thin thread, the central node is called E-domain, and consists of the association of the six N-terminal domains; E-domain is joined with the 2 lateral nodules called D-domain (the C-terminal domain of the Bβ and γ chains) by two coiled-coil regions that are the 3 chains Aα, Bβ and γ, that have a secondary α-helix structure, wrapped on themselves.
CELLULAR FUNCTIONS
Fibrinogen, called Coagulation Factor I, is a proenzimatic protein excreted by parenchimal hepatic cells. In circle, it’s converted into fibrin by thrombin to synthesize the soluble clot, that will be transformed into definitive clot by the XIII coagulation factor with the presence of Ca ++ ions.
Moreover, fibrinogen is also a cofactor of the platelet’s aggregation, in fact during the coagulation process it’s used by activated platelets as molecular bridge to form platelet’s aggregates.

REGULATION
Effects of triiodothyronine-induced hypermetabolism on factor 8 and fibrinogen in man. 1969
Triiodothyronine (liothyronine sodium) (400-500 μg/day for 14 days) was given to six normal subjects. Factor VIII (antihemophilic globulin) activity increased from 109 to 167% (P < 0.05); fibrinogen increased from 344 to 581 mg/100 ml (P < 0.01). To test whether the increases in factor VIII activity and fibrinogen were mediated by beta adrenergic receptors, propranolol (20 mg every 6 hr) was given orally to four other normal subjects in addition to triiodothyronine for 14 days. Factor VIII increased from 100 to 161%; fibrinogen increased from 374 to 564% (P < 0.01). Factor VIII activity did not change in a severe classical hemophiliac made hypermetabolic with triiodothyronine, but it increased from 39 to 82% in a patient with von Willebrand's disease. Triiodothyronine-induced hypermetabolism increased the incorporation of selenomethionine-75Se into plasma fibrinogen. These results suggest that the increases in clotting factor activity during triiodothyronine-induced hypermetabolism reflect an effect of increased protein synthesis rather than enhanced stimulation of beta adrenergic receptors.
Fibrinogen Disorders
Genetic
Quantitative Fibrinogen Disorders
The quantitative fibrinogen disorders belong to CID, Coagulation Inherited Disease, with a prevalence of 1-2:1.000.000 and depend on the presence of mutations that affect the fibrinogen genes. Afibrinogenaemia is a recessive autosomal disorder due to the presence of homozygous mutations o 2 mutations that affect the 3 genes in the same chromosome (phenomenon called “complex heterozygosis”). The patients affected have functional protein levels not registrable, extremely low or not registrable immunoreactive protein levels and can incur hemorrhagic episodes with variable severity.
Hypofibrinogenaemia is the state of heterozygosis of Afibrinogenaemia, and it is therefore a dominant autosomal disorder. The patients affected have a functional and immunoreactive protein level of fibrinogen under the minimum normal level, that is 160 mg/dl. Normally they are asymptomatic, but can incur hemorrhagic episodes.
Qualitative Fibrinogen Disorders
These disorders are defined by the presence of an abnormal variant of plasmatic fibrinogen (Dysfibrinogenaemia), usually associated with normal antigenic levels of protein, but in some cases they could present a reduction of the circulating protein (Hypo/Dysfibrinogenaemia).
The hereditary dysfibrinogenaemia is generally associated with an hereditary dominant autosomal pattern, with many heterozygous patients for the mutation causing the disease.
Acquired
High levels of fibrinogen could be due to hepatic hepatitis, atherosclerosis, rheumatoid arthritis, hepatic cirrhosis, viral hepatitis, gout, pregnancy, myocardial infarction, renal failure, multiple myeloma, oral contraceptive, traumas and burns.
Lower levels of fibrinogen could be due to prostate cancer, hemorrhagic events, phosphoric intoxication, acute infections, liver failure and use of anticoagulants.