Background
5-alpha-reductase type 2 deficiency (5-ARD) is an autosomal recessive sex-limited condition resulting in the inability to convert testosterone to the more physiologically active dihydrotestosterone (DHT).
Because DHT is required for the normal masculinization of the external genitalia in utero, genetic males with 5-alpha-reductase type 2 deficiency are born with ambiguous genitalia.
Patients with 5-alpha-reductase type 2 deficiency usually present with striking ambiguity of the genitalia, with a clitoral-like phallus, severely bifid scrotum, pseudovaginal perineoscrotal hypospadias, and a rudimentary prostate. Occasionally, patients can appear more masculinized; they may lack a separate vaginal opening, have a blind vaginal pouch that opens into the urethra, and have isolated penile hypospadias or even a penile urethra. The uterus and fallopian tubes are absent because of the normal secretion of the müllerian-inhibiting factor. Testes are intact and are usually found in the inguinal canal or scrotum; however, cryptorchidism is frequently described with testes occasionally located in the abdomen. Wolffian duct differentiation is normal with seminal vesicles, vasa differentia, epididymides, and ejaculatory ducts. The prostate is small, nonpalpable, and rudimentary in adulthood. Neither benign prostate hyperplasia (BPH) nor prostate cancer has been reported in these patients.
Pathophysiology
The root cause of this disorder is a deficiency in the 5-alpha-reductase type 2 isoenzyme, which transforms testosterone to DHT. DHT is a more potent androgen than testosterone and is bound selectively to the androgen receptors in genital skin and fibroblasts. The 5-alpha-reductase type 2 isoenzyme is expressed in external genital tissues early in gestation, making its action necessary for the development of normal male genital anatomy in the fetus. As with most single enzyme disorders, 5-alpha-reductase type 2 deficiency is autosomal recessive and sex limited because it only causes a clinically significant disorder in genetic males, with very subtle phenotypic changes in homozygous females.

Biochemical effects of 5-alpha-reductase type 2 deficiency in testosterone biosynthesis. Typically levels of testosterone are elevated, whereas levels of dihydrotestosterone (DHT) are significantly decreased, leading to male undervirilization.
Two genes coding for 5-alpha-reductase have been identified, each for a slightly different isoenzyme. The gene for 5-alpha-reductase type 2 has been determined to be on chromosome 2. Lack of expression of this gene clinically correlates with the symptoms of 5-alpha-reductase type 2 deficiency. In adulthood, the 5-alpha-reductase type 2 isoenzyme is expressed in high levels in the prostate, genital skin, epididymis, seminal vesicle, and liver.
The gene for 5-alpha-reductase type 1 has been located on chromosome 5. Its product is expressed only in nongenital skin and liver at low levels from the time the individual is aged 3 years until puberty, at which time enzyme expression is measurable in sebaceous glands and scalp. Linkage analysis has demonstrated that the type 1 enzyme is unrelated to the clinical syndrome of 5-alpha-reductase type 2 deficiency. Interestingly, partial virilization of males with 5-alpha-reductase type 2 deficiency occurs at puberty and may be attributable to the rise in type 1 enzyme activity at that time.
More than 33 different mutations of this gene have been reported in people with clinical and biochemical evidence of the enzyme deficiency. Correlation between the severity of the syndrome and a particular gene defect has not been observed.
Epidemiology
Although frequencies for various countries are not established, increased frequency is reported in the Dominican Republic, some highland tribes in New Guinea, and in Turkey. The high frequency in these areas represents the effect of consanguinity in specific kindreds.Overall, more than 50 families with this disorder have been described in several parts of the world. In a few patients with 46,XY disorders of sexual development (DSD) due to 5-alpha-reductase type 2 deficiency diagnosed by clinical and hormonal findings. In general, intersex conditions as a whole are uncommon, with an overall incidence of 1:5500.
Clinical 5-alpha-reductase type 2 deficiency is limited to genetic males. Although the enzyme deficiency can be documented in homozygous females, no clinical or developmental need for DHT is documented in women.
History
Diagnosis of 5-alpha-reductase type 2 deficiency (5-ARD) is usually made in the newborn period when the infant presents with ambiguous genitalia.No risk factors or clinical markers in pregnancy are known. Genital ambiguity is occasionally diagnosed prenatally when an infant who is demonstrated by amniocentesis or chorionic villus sampling to have XY karyotype fails to have a demonstrable penis on ultrasonography.
If 5-alpha-reductase deficiency is considered in a newborn, a broad approach to ambiguous genitalia should be taken.The utmost care should be taken not to assign a gender to the child before evaluation and consultation with an experienced, multidisciplinary team. The first words spoken to the parents are likely to be remembered and should focus on the overall health of the infant.
Initial evaluation should begin with a careful history.Assess prenatal and maternal past medical and family history. Specific questions should be asked regarding relatives with disorders of sexual development (DSDs), neonatal deaths, amenorrhea, or infertility and consanguinity. Parental consanguinity increases the child's risk for autosomal recessive disorders including 5-alpha reductase type 2 deficiency.
Physical
Workup of ambiguous genitalia must include careful physical examination to characterize the degree of virilization on the Prader scale , presence of palpable gonadal tissue, and overall health of the child.
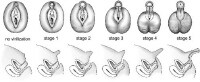
Prader scale reflecting the degree of virilization of the external genitalia. The internal genitalia reflect the changes in the urogenital sinus in response to the presence or absence of mullerian inhibiting hormone (MIH)
Phenotypic findings in a newborn are limited to the genitalia.The spectrum of findings ranges from minimal undervirilization presenting with normal male anatomy, except for isolated micropenis or hypospadias, to severe undervirilization presenting as normal female external genitalia with mild clitoral enlargement as the only physical finding.Most commonly, the external genitalia exhibit labial appearance to the labioscrotal folds with some mild rugation or pigmentation present in some patients.The phallus is indeterminate in size. Its length falls between 1 cm and 2 cm.The urethra may empty anywhere from the tip of the phallus to the perineum, with the latter observed more often.The testes are usually in the inguinal canals bilaterally; however, in some individuals with 5-alpha-reductase type 2 deficiency, the testes can be found in the labioscrotal folds or retained in the abdomen.A pseudovaginal blind-ending introitus is usually present with a normal hymen.Because the uterus is absent, rectal examination results are negative for a cervical mass. The rest of the examination findings are within normal limits.
Clear signs of virilization predominate at puberty. The escutcheon is male in distribution. The phallus exhibits definite enlargement. The shoulders are relatively broad and the hips are narrow. Muscularity and body hair may increase. No breast development is generally present. A prominent Adam's apple may start to develop. Facial hair develops. The child's voice may begin to deepen. The mucosa of the vaginal introitus remains atrophic in appearance (remaining red) rather than the thickened pink of estrogen-stimulated mucosa.
Medscape Medscape2