DEFINITION
GLUT4 is one of 13 sugar transporter proteins (GLUT1- GLUT12, and HMIT) encoded in the human genome that catalyzes hexose transport across cell membranes through an ATP-independent, facilitative diffusion mechanism. GLUT4 is highly espresse in adipose tissue and skeletal muscle. It displays the unique characteristic of a mostly intracellular disposition in the unstimulated state that is acutely redistributed to the plasma membrane in response to insulin and other stimuli (Regulated transport of the glucose transporter GLUT4, 2002) (Signaling Mechanisms That Regulate Glucose Transport, 1999).
GLUT4 is a protein of 509 aminoacid and a molecular wheight of 45 kDa.
THE GENE
The insulin-responsive glucose transporter 4 gene (SLC2A4) maps to 17p13. The gene is composed by 8000 bp and contains 11 exons.
CHEMICAL STRUCTURE AND IMAGES
GLUT4 contain 12-transmembrane domains (Figure below). GLUT4 contains unique sequences in its N- and COOH terminal cytoplasmic domains that direct its characteristic membrane trafficking capability (Figure below), these include a distinctive N-terminal sequence with a potentially critical phenylalanine residue (Roles of the N- and C-termini of GLUT4 in endocytosis, 2001) as well as dileucine and acidic motifs in the COOH terminus (Intracellular targeting and retention of the glucose transporter GLUT4 by the perinuclear storage compartment involves distinct carboxyl-tail motifs, 2000). These motifs likely govern kinetic aspects of both endocytosis and exocytosis in a continuously recycling trafficking system. These considerations place GLUT4 at the interface of two exciting fields of biology—insulin signaling and membrane trafficking.


the aminoacid more expressed in this transporter is arginine that through the NO synthase creates NO, an important vasodilator that enables entry of oxygen into the muscle cell. The presence of oxygen in muscle cells allows the catabolism of glucose.
SYNTHESIS
The relative abundance of GLUT4 in skeletal muscle depends on the developmental stage and the fibre type composition, GLUT4 is differentially expressed in oxidative and glycolytic muscle fibres.
Skeletal muscle expression of GLUT4 is low in the fetal period and a continuous induction of GLUT4 mRNA and protein takes place in the perinatal phase.
With respect to the mechanisms responsible for the transition of skeletal muscle GLUT4 expression during the prenatal phase, thyroid hormones, contractile activity and neural-derived trophic factors may play a role. Thus, congenital hypothyroidism markedly impairs GLUT4 protein and mRNA levels in skeletal and cardiac muscle. The effect thyroid hormone deficiency on GLUT4 expression is reversible and a single injection of T3 causes a marked and rapid increase in the levels of GLUT4 mRNA and protein in cardiac muscle. Thyroid hormones have also been implicated in the control of glucose transport in skeletal muscle in adulthood. Thus, long-term T3 administration stimulates GLUT4 expression and glucose transport in skeletal muscle (Role of calcium and AMP kinase in the regulation of mitochondrial biogenesis and GLUT4 levels in muscle, 2004).
Contractile activity and innervation regulates (through Ca2+ and other signals) the ratio of GLUT4 expression in skeletal muscle in an inverse manner.
The induction of GLUT4 occur at the end of fetal life and coincide with the timing of skeletal muscle innervation (nerve signal that induces increase in cytoplasmic calcium), the expression of GLUT4 is repressed in response to muscle denervation during adult life (Role of calcium and AMP kinase in the regulation of mitochondrial biogenesis and GLUT4 levels in muscle, 2004).
Muscle contraction increases GLUT4 expression. Physical training increases insulin-mediated whole-body glucose utilization in humans, which reflects an adaptation in skeletal muscle elicited by local contraction-dependent mechanisms. Physical training in humans does not alter insulin receptor function, but enhances the skeletal muscle content of GLUT4.
During muscle contraction ATP is hydrolysed to ADP by myosin ATPase to drive the cyclic association and dissociation of contractile proteins. Two reactions, driven by creatine kinase and adenylate kinase, act to replenish ATP by hydrolysing creatine phosphate and ADP to produce creatine and AMP respectively. Thus, during contractile activity of sufficiently high intensity ATP and creatine phosphate levels fall while the level of AMP rises. These changes in the levels of high-energy phosphates stimulate increases in GLUT4.
A myocyte enhancer factor 2 (MEF2) binding site in the glut4 promoter located between -466 and -457, relative to the transcription start, site has been proposed to be essential for the specific expression of GLUT4 in skeletal muscle cells. Disruption of this MEF2-binding site ablates tissue-specific GLUT4 expression (Role of calcium and AMP kinase in the regulation of mitochondrial biogenesis and GLUT4 levels in muscle, 2004).
There is mounting evidence that the increase in cytosolic Ca2+ (muscle contraction and innervation) triggers the events that lead to increased GLUT4 expression.
That increases in cytosolic Ca2+, mediated either by intermittent release of Ca2+ from the sarcoplasmic reticulum or Ca entry through the sarcolemma, stimulate GLUT4 expression and that these effects are not mediated by a decrease in high-energy phosphate. Furthermore, in support of the role of Ca, dantrolene, in concentrations that inhibit most of the increase in cytosolic Ca2+, also inhibits the increases in GLUT4 proteins. Data showing increases in PGC-1, NRF-1 and NRF-2, and mitochondrial transcription factor A, in response to raising cytosolic Ca2+, provide evidence that the same mechanisms that mediate increases in GLUT4 in response to exercise also increase GLUT4 in Ca2+ stimulated myotubes (Role of calcium and AMP kinase in the regulation of mitochondrial biogenesis and GLUT4 levels in muscle, 2004).
CELLULAR FUNCTIONS
Elevated glucose levels are rapidly returned to normal (5–6 mM) even after huge caloric ingestions, and they are maintained at only slightly lower levels during longterm starvation. Such control prevents severe dysfunctions such as loss of consciousness due to hypoglycemia and toxicity to peripheral tissues in response to the chronic hyperglycemia of diabetes. The major cellular mechanism for disposal of an exogenous glucose load is insulin-stimulated glucose transport into skeletal muscle. Skeletal muscle both stores glucose as glycogen and oxidizesit to produce energy following the transport step. The principal glucose transporter protein that mediates this uptake is one isoform (Gene name, SLC2A4; protein name: GLUT4) of a family of sugar transporter proteins. The GLUT4 glucose transporter is thus a major mediator of glucose removal from the circulation and a key regulator of whole-body glucose homeostasis.
Under normal conditions, this carrier is located in the cytoplasmic vesicles and its translocation to the cell membrane is stimulated by the binding of insulin receptor membrane. This process facilitates the movement of glucose from the interstitial fluid within the cell. When the blood glucose concentration returns to normal and the insulin is removed, the molecules of GLUT4 are slowly removed from the plasma membrane by endocytosis and sequestered in intracellular vesicles.
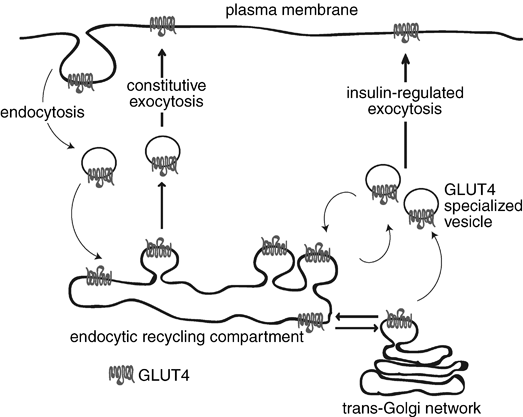
REGULATION
A central role for GLUT4 in whole-body metabolism is strongly supported by a variety of genetically engineered mouse models where expression of the transporter is either enhanced or ablated in muscle. The heterozygous GLUT4+/- mice that display decreased GLUT4 protein in muscle, show the expected insulin resistance and propensity toward diabetes that is consistent with a major role of GLUT4 in glucose disposal (Reduced glucose uptake precedes insulin signaling defects in adipocytes from heterozygous GLUT4 knockout mice, 2000).
The profound effects on whole-body glucose homeostasis observed in mouse models of GLUT4 deficiency or overexpression heighten the potential physiological importance of changes in endogenous GLUT4 expression in different states. Examples of particular interest are the the upregulation of GLUT4 expression in skeletal muscle in response to exercise or thyroid hormone, also, GLUT4 expression is greatly decreased in muscle atrophy and denervation consistent with decreased energy requirements in these conditions.
Newly synthesized and glycosylated GLUT4 enters a continuously recycling pathway that concentrates most of the GLUT4 in intracellular membrane systems in unstimulated muscle cells.
Both insulin and exercise acutely stimulate GLUT4 recruitment to the cell surfaces of muscle independent of transcription or translation. Nevertheless, these two physiological stimuli initiate distinct signaling mechanisms that lead to enhanced GLUT4 translocation and glucose uptake.
The canonical insulin signaling pathway is triggered by activation of the insulin receptor (IR) tyrosine kinase leading to tyrosine phoshphorylation of insulin receptor substrate proteins (IRS) and their recruitment of PI 3-kinase, which catalyzes conversion of phosphatidylinositol (4,5)P2 to phosphatidylinositol (3,4,5)P3 (denoted PIP3). PIP3 in turn triggers the activation of the protein kinase Akt through the actions of two intermediate protein kinases, PDK1 and Rictor/mTOR. The GTPase activating protein TBC1D4, denoted AS160, is a substrate of Akt that catalyzes inactivation of Rab proteins 2A, 8A, 10 and 14. Rab proteins are critical organizers of intracellular membrane trafficking. Expression of mutant AS160 lacking Akt-specific phosphorylation sites inhibits insulin-stimulated GLUT4 translocation (Insulin-stimulated Phosphorylation of a Rab GTPase-activating Protein Regulates GLUT4 Translocation, 2003), suggesting it is a negative regulator that is itself inhibited by insulin through Akt.
Muscle contraction also increases AS160 phosphorylation, but it may do so by a PI 3-kinase independent mechanism. Contraction-induced AS160 phosphorylation is mediated through the AMPK pathway, providing a potential convergence of insulin and exercise-mediate signaling to GLUT4.
Two cellular consequences of muscle contraction, a transient increase in intracellular calcium concentration ([Ca2+]i) and an increase in [AMP]/[ATP] ratio, are thought to contribute to enhanced GLUT4 translocation to the cell surface. The former signal is mediated through activations of the protein kinase CaMKII and perhaps conventional protein kinase C. In addition, Ca2+/CaM appears to activate the AMPK signaling pathway through upstream kinases CaMKK α and β. AMPK acts as a cellular energy gauge that is allosterically activated by increasing [AMP]/[ATP] ratios and perhaps other mechanisms. This transforms AMPK into a better substrate for at least one of its upstream activator kinases, e.g., LKB1, or a potentially poorer substrate for one of its phosphatases.
Muscle glycogen appears to be potent negative regulator of AMPK activity, thus providing a negative feedback mechanism for AMPK-mediated glucose uptake. The critical substrates (‘‘effectors’’ in Figure below) that mediate GLUT4 translocation downstream of these protein kinases are unknown.

Insulin markedly stimulates GLUT4 exocytic pathways while also significantly inhibiting its endocytosis from the plasma membrane, which together cause the overall redistribution of GLUT4 to the cell surface. That endocytosis of GLUT4 in unstimulated cells occurs mostly through a cholesterol dependent, clathrin adaptor AP-2-independent pathway, while insulin selectively inhibits this. Thus, GLUT4 recycling in insulin-stimulated muscle may be a specialized case of the clathrin-dependent recycling pathway which occurs in all cells, best studied in reference to the transferrin receptor.
ALTERATION IN INSULIN RESISTANCE
Insulin resistance is strongly associated with type II diabetes. “Diabetogenic” factors including FFA, TNFalpha and cellular stress induce insulin resistance through inhibition of IRS1 functions. Serine/threonine phosphorylation, interaction with SOCS, regulation of the expression, modification of the cellular localization, and degradation represent the molecular mechanisms stimulated by them. Various kinases (ERK, JNK, IKKbeta, PKCzeta, PKCtheta and mTOR) are involved in this process. The development of type II diabetes requires impaired beta-cell function. Chronic hyperglycemia has been shown to induce multiple defects in beta-cells. Hyperglycemia has been proposed to lead to large amounts of reactive oxygen species (ROS) in beta-cells, with subsequent damage to cellular components including PDX-1. Loss of PDX-1, a critical regulator of insulin promoter activity, has also been proposed as an important mechanism leading to beta-cell dysfunction. Although there is little doubt as to the importance of genetic factors in type II diabetes, genetic analysis is difficult due to complex interaction among multiple susceptibility genes and between genetic and environmental factors. Genetic studies have therefore given very diverse results. Kir6.2 and IRS are two of the candidate genes. It is known that Kir6.2 and IRS play central roles in insulin secretion and insulin signal transmission, respectively.
In the insulin resistance the exocytose of GLUT4 insulin-stimulated, in skeletal muscle, is impaired because the inhibition of IRS1 functions blocks the insulin’s pathways.
Other reasons for the failure of GLUT4 exocytosis may be the presence of conditions impairing cytoplasmic vesicles movement (lack of calcium or iron overload eg).
This causes a constant hyperglycemia that lead to a worse diabetes, because the skeletal muscle is the most tissue that regulates the glucose homeostasis and its impairement is very dangerous for the patient health.
Moreover insulin-stimulated AS160 phosphorylation is impaired in skeletal muscle of type II diabetic patients (Insulin-Stimulated Phosphorylation of the Akt Substrate AS160 Is Impaired in Skeletal Muscle of Type 2 Diabetic Subjects, 2005).
Therefore this pathways are important in type II diabetes and the study of them would be important and useful for type II diabetes therapy.