Definition
Imatinib mesylate is currently marketed by Novartis as Gleevec in USA and Glivec in Europe.
Designated chemically as 4-[(4-Methyl-1-piperazinyl)methyl]-N-[4-methyl-3-[[4-(3-pyridinyl)-2-pyrimidinyl]amino]-phenyl]benzamide methanesulfonate, it is also called CGP57148B or STI571.
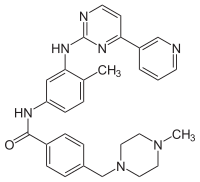
Paradigm of targeted molecular therapy, its structural formula is:
Classification
1. Antineoplastic agent
2. Protein kinase inhibitor/antagonist
Indications
- bcr-abl
- Newly Diagnosed Philadelphia Positive Chronic Myeloid Leukemia (Ph+ CML)
- Ph+ CML in Blast Crisis, Accelerated Phase or Chronic Phase after Interferon-alpha therapy
- Pediatric Patients with Ph+ CML in Chronic Phase
- Ph+ Acute Lymphoblastic Leukemia
- pdgfrB/pdgfrA
- Myelodysplastic/Myeloproliferative Diseases
- fIp1l1-pdgfrA
- Aggressive Systemic Mastocytosis
- Hypereosinophilic Syndrome and/or Chronic Eosinophilic Leukemia
- col1a1-pdgfrB
- Dermatofibrosarcoma Protuberans
- kit+
- Kit+ Gastrointestinal Stromal Tumors (GIST)
- Adjuvant Treatment of GIST
Pharmacokinetics
Absorption:
- oral administration (Cmax achieved within 2-4 hours post-dose).
- bioavailability 98%.
Distribution:
at clinically relevant concentrations of imatinib, binding to plasma proteins is approximately 95%, mostly due to albumin and alpha1-acid glycoprotein.
Metabolism and interactions:
imatinib is primarily metabolized by CYP3A4 to an active metabolite, an N-demethylated piperazine derivative (CGP 74588). Other cytochrome P450 isoforms, such as CYP1A2, CYP2D6, CYP2C9, and CYP2C19, play a minor role in its metabolism.
Imatinib is a potent competitive inhibitor of CYP2C9, CYP2D6, and CYP3A4: it may interact with different drugs and different drugs may affect its levels.
Imatinib can increases effect of:
- Acenocoumarol, dicumarol, warfarin and anisindione (anticoagulant agents), CYP2C9 substrates: imatinib inhibits CYP2C9, then can increase drugs concentrations.
- Atorvastatin, lovastatin and simvastatin (ipocholesterolemic agents), CYP3A4 substrates: imatinib inhibits CYP3A4, then can increase drugs concentrations.
- Nifedipine (Ca2+ channel blockers), CYP3A4 substrate: imatinib inhibits CYP3A4, then can increase drug level.
- Pimozide (antipsychotic agents), CYP3A4 substrate: imatinib inhibits CYP3A4, then can increase drug level.
- Cyclosporine (immunosupressant agents), CYP3A4 substrate: imatinib inhibits CYP3A4, then can increase drug level.
- Metoprolol (antihypertensive agents), CYP2D6 substrate: imatinib inhibits CYP2D6, then can increase drug level.
Drugs that may affect imatinib levels:
1. Decrease (CYP3A4 inducers):
- Carbamazepine
- Dexamethasone
- Hydantoin
- Phenobarbital
- Rifampin
- Primidone
- Hypericum
CASE REPORT: IMATINIB-RIFAMPIN INTERACTION
Rifampin is a potent inducer of cytochrome P-450 hepatic enzyme system, especially isoform 3A4 (CYP3A4).
Male, age 79.
June 2008: diagnosis of TBC. Immediately starting treatment with rifampicin.
Concomitant therapy with Glivec® (300 mg/day) for previous CML diagnosis.
Patient undergoes test for imatinib plasma level determination, in order to verify if concentration obtained was efficacy in terms of CML treatment. It's known from literature that imatinib plasma concentration should reach at least 1000 ng/ml to be effective (Picard et al, 2007, Larson et al, 2008).
Because of drugs interaction, imatinib plasma level decreases a lot with the consequence of less effective treatment in terms of molecular and cytogenetic response.
At first control, in july, imatinib plasma level was around 450 ng/ml and in august, for a second control, level was even lower: less than 300. In september 2008 patient completes antibiotic therapy. In one month imatinib plasma level increases a lot, till reaching 900 ng/ml. During concomitant rifampin administration, mean imatinib concentration decreased by 57%, due probably to the considerable increase in imatinib biotransformation.
In the same plasma samples it was performed quantification of rifampin level. It resulted in the expected therapeutic ranges: for tuberculosis patient was, then, suitably treated.
Today, patient, followed by Ematology of San Luigi Hospital (Orbassano, Turin) is still under imatinib therapy. Imatinib plasma concentrations range from 900 to 1200 ng/ml. (No CYP3A4 inducers anymore!)
2. Increase (CYP3A4 inhibitors):
- Antibiotics (clarithromycin, erythromycin and josamycin)
- Antifungal agents (itraconazole and ketoconazole)
CASE REPORT: IMATINIB-VORICONAZOLE INTERACTION
Voriconazole inhibits metabolic activity of CYP2C19, CYP2C9 and CYP3A4.
Female, age 57.
April 2009: diagnosis of pulmonary mycosis. Immediately starting treatment with voriconazole.
Concomitant therapy with Glivec® (300 mg/day) for previous CML diagnosis.
Patient undergoes test for imatinib plasma level determination, in order to verify if concentration obtained was efficacy in terms of CML treatment. It's known from literature that imatinib plasma concentration should reach at least 1000 ng/ml to be effective (Picard et al, 2007, Larson et al, 2008).
Because of drugs interaction, imatinib plasma level increases a lot with the consequence of emerging adverse haematological reactions (grade 2).
At control for imatinib plasma level, determination was 3300 ng/ml. After stopping antifungal treatment imatinib level decreased a lot in a few weeks, arriving around 2100 ng/ml. During concomitant voriconazole administration, an increase in imatinib level (36%) was recorded, due to the remarkable reduction of imatinib biotransformation.
In the same plasma samples it was performed quantification of voriconazole level. It resulted in the expected therapeutic ranges: for mycosis patient was, then, suitably treated.
Today patient, followed by Ematology of San Luigi Hospital (Orbassano, Turin) is still under imatinib therapy. Imatinib plasma concentrations range from 1000 to 1500 ng/ml. (No CYP3A4 inhibitors anymore!)
Analysis of two case reports suggests that a loss of efficacy in CML treatment or severe drug reactions to imatinib may be related not only to administration of too low or too high doses, respectively, but also to pharmacokinetic interactions with CYP3A4 inducers/inhibitors. Monitoring of imatinib plasma concentrations, then, may be helpful for identifying different situations.
Cases like these may happen every days, not only for co-administration of drugs with imatinib, but, of course, in different fields of therapy. To ensure correct therapy for patients, useful should be know potential drug-drug interactions as much as possible, but free database on-line not fully help us!
There are any drugs databases available as drugbank, toxnet and medlineplus but not always report informations about drugs interactions.
Interesting link for consultation is CYP450 table
Elimination:
elimination half-lives are 18 hours for imatinib and 40 hours for CGP 74588. Imatinib is eliminated predominately in the feces, mostly as metabolites. Approximately 81% of the dose is eliminated within 7 days in the feces (68%) and urine (13%). Unchanged imatinib accounts approximately for 25% of the dose (5% urine, 20% feces).
Molecular mechanism
Imatinib is a protein-tyrosine kinase inhibitor that blocks selectively a number of tyrosine kinase (RTK) enzymes. It occupies the RTK active site, leading to a decrease in its activity. Imatinib is specific for the TK domain in bcr-abl, the constitutive abnormal RTK created by Philadelphia chromosome in CML. Imatinib competitively inhibits the binding of ATP to the ATP binding pocket of bcr-abl. This results in inhibition of proliferation and induction of apoptosis in bcr-abl positive cells, with no effect on normal cells.
Below bcr-abl kinase, which causes CML, inhibited by imatinib (red small molecule).

Below imatinib mechanism of action.

In the case of GIST, imatinib interrupts KIT-mediated signal transduction. Imatinib is also an inhibitor of the receptor RTK for platelet-derived growth factor (PDGF) and stem cell factor (SCF) and inhibits PDGF- and SCF-mediated cellular events.

Pharmacogenetics
LINKS
Toxicity
1. Severe Congestive Heart Failure and Left Ventricular Dysfunction: occasionally, less than 1%.
2. Hepatic: occasionally severe.
3. Hemorrhage (grade 3/4): 1.8% CML, 12.9% GIST.
4. Gastrointestinal Disorders: rare.
5. Dermatologic: rare.
Side effects
1. Fluid Retention and Edema (4% CML, 10% GIST).
2. Hematologic (anemia, neutropenia and thrombocytopenia grade 3/4) during first month of therapy.
3. Vomiting, diarrhea, loss of appetite.
4. Dry skin, hair loss.
5. Swelling (especially in the legs or around the eyes).
5. Muscle cramps.
Resistance
The mechanism of resistance to imatinib appears to be due a variety of factors, including BCR-ABL gene amplification, mutations in the protein that could alter binding, or over-expression of transporters.
Two second-generation bcr-abl inhibitors, dasatinib and nilotinib, active against most of imatinib resistant bcr-abl mutants, have recently been tested in clinical trials (Shah et al, 2003, Gorre et al, 2001, Lombardo et al, 2004, Kantarjian et al, 2006, Cortes et al, 2007). At present imatinib pharmacokinetics has been well investigated, while only limited information on dasatinib and nilotinib are available. Very recent studies reported even comparison between imatinib and nilotinib, showing that imatinib is a more potent inhibitor of BCR-ABL than nilotinib. (Saglio et al, 2010).